【共识】中国POEMS综合征周围神经病变诊治专家共识
发表人:
韩小磊
阅读量:
684人
POEMS综合征是一种病因和发病机制不清的、罕见的多系统疾病,主要表现(按照字母顺序)为:P:多发性神经病变(包括四肢麻木无力,以下肢远端无力为主);O:器官肿大(包括肝脾大、淋巴结肿大,淋巴结活体组织病理检查常为Castleman病表现);E:内分泌异常(包括性功能减退、甲状腺功能减退、肾上腺皮质功能不全、糖尿病等);M:血清中存在M蛋白(经蛋白电泳或免疫固定电泳证实,一般都为IgG或IgA-λ型);S:皮肤改变(皮肤颜色变黑变硬、体毛增多变硬);其他表现还有腹腔积液、胸腔积液和水肿、肺动脉高压、视乳头水肿等。本病于1956年首先由Crow[1]描述,1968年随后由Fukase描述,Nakanishi等[2]将其称为Crow-Fukase综合征。Takatsuki和Sanada[3]首先确认并全面描述本病,因此也有人称为Takatsuki综合征。Bardwick在1980年首次将主要症状的首字母组合,形成了现在的POEMS综合征[4]。几乎所有病例都合并浆细胞增生性疾病,最常见为骨硬化性骨髓瘤,其次为髓外浆细胞瘤,溶骨性多发性骨髓瘤少见。病例多合并内分泌功能紊乱、心力衰竭和恶病质。目前来自法国、美国、中国和日本的小样本的流行病学调查,显示该病患病率约为0.3/10万[5,6,7]。POEMS综合征患者均有周围神经受损,甚至有不少患者因首发症状为周围神经病表现而就诊神经内科,因此,在中华医学会神经病学分会领导下,中华医学会神经病学分会周围神经病协作组、肌电图与临床神经电生理学组和神经肌肉病学组专家共同合作编写该共识,以帮助临床医生规范诊治POEMS综合征患者。
一、临床特点
1.多发性周围神经病:
疾病初期常见为隐袭起病的渐进性的运动感觉周围神经病。为双下肢起病,逐渐向上发展。通常伴有麻木、刺痛和发凉感,随后出现无力症状,初期无力症状不重,主诉一般为难以上楼、难以站起。随病程进展,运动症状较感觉症状突出。除视乳头水肿外不累及脑神经。体检表现为双下肢远端为重的感觉周围神经病和上下肢远端无力及肌肉萎缩的周围神经病,通常对称存在。病程较为良性,但致残率高,患者生活质量较差。100%的POEMS综合征并发多发性周围神经病,这也是诊断POEMS的首个条件。
2.其他表现:(1)脏器肿大,通常为肝脾肿大,淋巴结肿大;(2)内分泌异常,表现为甲状腺功能减退、性功能减退、肾上腺皮质功能不全、糖尿病等表现;(3)M蛋白,通常为λ型;(4)皮肤改变(皮肤颜色变黑变硬,体毛增多等)。推荐意见:(1)对慢性进展的周围神经病患者,应该注意询问和检查是否存在脏器肿大、内分泌异常、皮肤改变,如果存在这些异常,应该考虑POEMS综合征的可能。(2)POEMS综合征并发周围神经病达100%。
二、实验室检查
1.神经传导检测和针极肌电图:上下肢运动、感觉神经传导速度(包括正中、尺、桡、腓总、胫、腓肠等周围神经)可见一条或多条神经运动末端潜伏期延长或神经传导速度(NCV)轻度减慢或减慢,或伴随有波幅下降[8,9]。针电极可见到周围神经损伤区域的肌肉失神经和神经源性损害。神经传导速度和针极肌电图为诊断POEMS周围神经病的损伤范围及程度的客观指标。
2.腰椎穿刺:
脑脊液可出现压力轻度增高或正常,细胞计数正常或轻度增高,蛋白定量常增高的细胞-蛋白分离现象但无特异性。腰椎穿刺在本病可以鉴别感染性或恶性细胞增殖性疾病[10]。
3.血生化检查:(1)血清或尿免疫固定电泳发现游离轻链,且M蛋白为IgG或IgA λ型;(2)骨放射检查:发现单个或多个骨髓破坏性病灶;(3)骨髓活体组织检查:半数患者可见浆细胞轻度增多(2%~5%),合并骨髓瘤者的轻链限制性浆细胞比例明显增高(>10%);(4)血浆或血清血管内皮生长因子水平检测,血浆>200 pg/ml或血清>1 920 pg/ml[11];(5)内分泌检查提示甲状腺,肾上腺皮质功能减退和血糖升高等;(6)超声提示肝脾肿大和淋巴结肿大。
4.腓肠神经活体组织检查:可见不同程度的节段性脱髓鞘和(或)轴索变性,以轴索变性和神经外膜的新血管生成[12]为主要表现。本病腓肠神经活体组织检查在高度怀疑淀粉样变和血管炎性周围神经病时有鉴别意义,但并非POEMS周围神经病诊断所必需。推荐意见:(1)神经传导和肌电图检查可帮助判断多发性周围神经病的性质和程度;(2)对于原因不明的慢性周围神经病,血清和尿免疫固定电泳发现λ型轻链可以作为首选的辅助检查;(3)脑脊液检查和血清免疫电泳除外其他病因。
三、诊断
临床表现为亚急性或慢性进行性多发性运动感觉周围神经病的患者在经过电生理诊断后需要进行常规的周围神经病的病因筛查,如血清或尿免疫固定电泳发现游离轻链,则应进行骨髓检查,以除外浆细胞增殖性疾病。如患者伴有下列症状或体征即可考虑POEMS的诊断:(1)不明原因的肝脾肿大;(2)不明原因的腹腔积液、胸腔积液或水肿;(3)不明原因的性功能减退或伴有甲状腺功能减退;(4)不明原因的皮肤变黑。2003年国际骨髓瘤协作组(The International Myeloma Working Group)发表了POEMS诊断标准,并于2007年做了修订[13],在2017年做了更新[14]。其中诊断的必要条件包括2条强制标准和至少1条主要标准和至少1条次要标准(表1)。
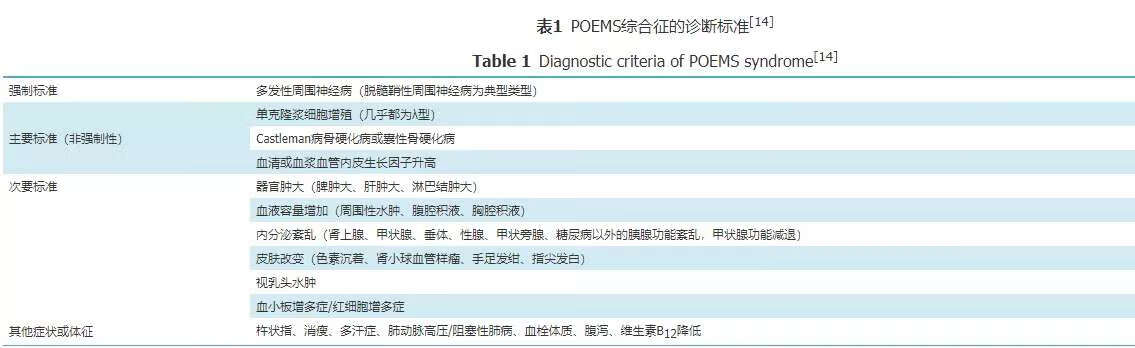
推荐意见:POEMS综合征的诊断条件和流程:慢性或亚急性周围神经病患者,如血清或尿免疫固定电泳发现游离轻链则应考虑POEMS的可能;伴有下列中的1条即可诊断POEMS综合征:(1)Castleman病;(2)骨硬化病或囊性骨硬化病;(3)血清或血浆血管内皮生长因子升高,和下列中的1条:①器官肿大(脾肿大、肝肿大、淋巴结肿大);②血液容量增加(周围性水肿、腹腔积液、胸腔积液);③内分泌紊乱(肾上腺、甲状腺、垂体、性腺、甲状旁腺、糖尿病以外的胰腺功能紊乱,甲状腺功能减退);④皮肤改变(色素沉着、肾小球血管样瘤、手足发绀、指尖发白);⑤视乳头水肿;⑥血小板增多症/红细胞增多症。
四、鉴别诊断
1.慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculo- neuropathy, CIDP):
早期慢性或亚急性进展性周围神经病易被误诊为CIDP,患者常随着系统病变的发现而诊断为POEMS综合征。CIDP患者不会出现POEMS的异常M蛋白、血管内皮生长因子升高,骨放射检查以及皮肤的改变可以区分两者。在神经电生理诊断中,POEMS综合征患者的周围神经病更常见到近端NCV减慢,CIDP患者更多见传导阻滞[15]。
2.单克隆丙种球蛋白病合并周围神经病,也称为副蛋白血症周围神经病(paraproteinemic
peripheral neuropathy):
在原因未明的特发性周围神经病患者中,有10%合并单克隆丙种球蛋白病,而意义未明单克隆球蛋白血症(monoclonal gammopathy of undetermined significance,MGUS)中有29%~71%合并周围神经病,提示单克隆丙种球蛋白病与周围神经病有关。患者同时存在两组临床症状、体征,即单克隆丙种球蛋白病所导致的多系统病变表现,以及周围神经受损害出现的周围性运动、感觉自主神经功能障碍表现。此为本病的临床特征之一。
MGUS或良性单克隆丙种球蛋白病合并周围神经病主要见于50岁以上,起病隐袭,临床表现为足麻木、感觉异常、平衡障碍和步态不稳,深感觉和触觉受累明显。半数患者有疼痛不适。约50%患者人抗髓鞘相关糖蛋白抗体阳性。MGUS虽然出现M蛋白,但不会出现血管内皮生长因子升高、骨放射检查以及皮肤的改变。
3.淀粉样变性周围神经病(amyloidotic peripheral neuropathy):
即周围神经的淀粉样变性,是淀粉样物质在周围神经沉积引起的一组严重的进行性感觉、运动周围神经病,伴自主神经功能障碍。本组疾病主要包括家族性淀粉样变性周围神经病、原发性轻链淀粉样变性、继发性淀粉样变性等。淀粉样变性周围神经病老年期发病,主要临床特征为小纤维神经病和自主神经功能低下。表现为痛性感觉异常,自主神经功能低下可在患病初期出现。其他系统受累时可有肝脾肿大、蛋白尿或肾病、异常球蛋白血症和巨舌,血或尿免疫电泳也可以合并M蛋白。骨髓活体组织检查结果不会出现POEMS综合征的异常增殖。NCV提示上下肢周围神经运动感觉均受累的周围神经病变,运动神经波幅和(或)感觉神经波幅降低伴有速度减慢,腓肠神经活体组织检查可见到偏振光苹果绿染色阳性或刚果红染色阳性,可以诊断。
推荐意见:(1)对于慢性多发性周围神经病,血或尿中出现异常增殖轻链(M蛋白)可以与CIDP和MGUS鉴别,但淀粉样变可以出现M蛋白;(2)骨髓活体组织检查结果阴性以及腓肠神经(或唇部、腹部脂肪)活体组织检查发现淀粉染色阳性可以帮助诊断淀粉样变。
五、治疗
1.POEMS综合征的治疗原则:
仅有骨髓受累(2个以内病灶)而未发现克隆性浆细胞病的患者推荐观察并每3~6个月评估。单纯骨髓病变数目大于或等于3个,或已发现克隆性浆细胞病证据的患者进行系统治疗,可根据病情和实际医疗条件选择:马法兰联合地塞米松或糖皮质激素单独应用、环磷酰胺联合地塞米松、自体造血干细胞移植、沙利度胺联合地塞米松、硼替佐米、贝伐单抗[16]等。
①病因治疗:对明确诊断的POEMS综合征进行针对性治疗;②神经营养修复治疗:临床可选择多种B族维生素(如硫胺素和甲钴胺等)针对神经营养修复治疗;③对症治疗:对伴有神经痛的患者可以给予治疗神经痛的药物改善生活质量,目前根据证据级别可以选择三环类抗抑郁药、钙离子通道调节剂、5-羟色胺和去甲肾上腺素再摄取抑制剂等[17]。在可作出POEMS综合征诊断的中心可以采用NCV和针极肌电图的变化作为客观评价神经病变的指标。
3.支持治疗:
对所有患者均需要血液科、神经科、康复科、内分泌科、肾内科、放射治疗科的多科协作。对患者除了治疗浆细胞病或骨髓瘤,还需要支持治疗,包括针对周围神经病的康复治疗和器械辅助、内分泌的替代治疗、浆膜渗出导致的水负荷过多应用利尿剂治疗以及针对血液高凝状态的抗血小板治疗等。
推荐意见:
(1)对临床诊断为POEMS综合征的患者需要血液科、神经科、康复科的联合治疗,以血液科治疗克隆性浆细胞病为病因治疗;(2)周围神经病的治疗在于早期诊断和神经营养治疗和对症治疗。
六、预后
POEMS综合征患者的预后较多发性骨髓瘤好。POEMS综合征患者发病中位年龄为51岁,进展缓慢,中位生存期为97个月,5年生存率为60%。马法兰(melphalan)治疗和肾功能正常有助于延长生存期[18]。神经病变的不断恶化是POEMS综合征的常见结局和死因,而继发于疾病进展和化疗后的骨髓衰竭是多发性骨髓瘤的常见死因。患者主要死于疾病进展、肺炎、脓毒血症、卒中、急性髓细胞白血病和多发性骨髓瘤。

本文由作者上传,文章内容仅供参考。如有相关事宜请联系jdh-hezuo@jd.com
本站内容仅供医学知识科普使用,任何关于疾病、用药建议都不能替代执业医师当面诊断,请谨慎参阅。
相关文章
文章 指南共识 | 中国重症肌无力诊断和治疗指南(一、诊断与鉴别诊断)
重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经-肌肉接头(neuromuscular junction,NMJ)传递障碍的自身免疫性疾病。乙酰胆碱受体(acetylcholine receptor,AChR)抗体是最常见的致病性抗体;此外,针对突触后膜其他组分,包括肌肉特异性受体酪氨酸激酶(muscle-specific receptor tyrosine kinase,MuSK)、低密度脂蛋白受体相关蛋白4(low-density lipoprotein receptor-related protein 4,LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR功能及NMJ信号传递。目前,MG的治疗仍以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(intravenous immunoglobulins,IVIG)、血浆置换(plasma exchange,PE)以及胸腺切除为主。MG全球患病率为(150-250)/百万,预估年发病率为(4-10)/百万。我国MG发病率约为0.68/10万,女性发病率略高;住院死亡率为14.69‰,主要死亡原因包括呼吸衰竭、肺部感染等。各个年龄阶段均可发病,30岁和50岁左右呈现发病双峰,中国儿童及青少年MG(juvenile myasthenia gravis,JMG)患病高达50%,构成第3个发病高峰;JMG以眼肌型为主,很少向全身型转化。最新流行病学调查显示,我国70-74岁年龄组为高发人群。 近年来,在MG诊疗方面取得了众多进展,积累了更多循证医学证据。为此,中国免疫学会神经免疫分会基于近5年国内外文献中的最新证据,参考相关国际指南,反复讨论,在对中国MG诊治指南(2015)更新修订的基础上编写了本指南。 新指南采用MGFA临床分型替代Osserman分型,旨在对疾病严重程度进行量化评估;提出MG亚组分类,指导精准化治疗;对治疗目标进行了定义;针对胸腺切除,利妥昔单抗、依库珠单抗等生物制剂的应用,眼肌型MG(ocular MG,OMG)早期免疫抑制治疗以及免疫检查点抑制剂(immune check point inhibitors,ICIs)治疗相关MG等方面提出了新的建议。 一、MG临床表现、分型及亚组分类 1.1 临床表现 全身骨骼肌均可受累,表现为波动性无力和易疲劳性,症状呈“晨轻暮重”,活动后加重、休息后可减轻。眼外肌最易受累,表现为对称或非对称性上睑下垂和/或双眼复视,是MG最常见的首发症状,见于80%以上的MG患者。面肌受累可致眼睑闭合无力、鼓腮漏气、鼻唇沟变浅、苦笑或呈肌病面容。咀嚼肌受累可致咀嚼困难。咽喉肌受累可出现构音障碍、吞咽困难、鼻音、饮水呛咳及声音嘶哑等。颈肌受累可出现抬头困难或不能。肢体无力以近端为著,表现为抬臂、梳头、上楼梯困难,感觉正常。呼吸肌无力可致呼吸困难。发病早期可单独出现眼外肌、咽喉肌或肢体肌肉无力;脑神经支配肌肉较脊神经支配肌肉更易受累。肌无力常从一组肌群开始,逐渐累及到其他肌群,直到全身肌无力。部分患者短期内病情可出现迅速进展,发生肌无力危象。 1.2 美国重症肌无力基金会(MGFA)临床分型 旨在评估疾病严重程度,指导治疗及评估预后。疾病严重程度可根据定量MG评分(quantitative MG score,QMGS)评估。 1.3 MG亚组分类及临床特点 MG临床表现具有极大异质性,以血清抗体及临床特点为基础的亚组分类,对MG个体化治疗及预后评估更具指导意义。 1.3.1 OMG:MGFA I型,可发生于任何年龄阶段。 我国儿童及JMG以眼肌型为主,很少向全身型转化。 成人发病的OMG,在眼肌症状出现2年内容易向全身型转化,亚裔人群2年自然转化率为23%-31%,低于西方人群(50%-80%);合并胸腺瘤、异常重复神经电刺激(RNS)结果、AChR抗体阳性、病情严重的OMG更易发生转化。 早期免疫抑制治疗减少OMG继发转化,部分儿童及青少年OMG可能会自行缓解。 1.3.2 AChR-全身型MG(generalized MG,GMG):该类患者血清AChR抗体阳性,无影像学怀疑或病理确诊的胸腺瘤;依据发病年龄可分为早发型MG(early-onset myasthenia gravis,EOMG)及晚发型MG(late-onset myasthenia gravis,LOMG)。EOMG是指首次发病在50岁之前,女性发病略高于男性,常合并胸腺增生,胸腺切除可获益,与HLA-DR3、HLA-B8以及其他自身免疫性疾病风险基因相关;LOMG是指首次发病在50岁以后,男性发病略高于女性,胸腺萎缩多见,少数伴胸腺增生的患者胸腺切除可能获益。 1.3.3 MuSK-MG:大约在1%-4%的MG患者血清中可检测到MuSK抗体,与AChR抗体(IgG1和IgG3)不同,绝大多数MuSK抗体属于IgG4亚型,其与AChR-IgG极少同时出现。MuSK-MG受累肌群较局限,以球部、颈部及呼吸肌受累为主,其次为眼外肌、四肢肌,主要表现为球麻痹、面颈肌无力。MuSK-MG与HLA-DQ5相关,通常不伴胸腺异常。 1.3.4 LRP4-MG:在1%-5%的MG以及7%-33%的AChR、MuSK抗体阴性MG患者可检测出LRP4抗体。LRP4-MG的临床特点尚不完全明确,有研究表明该亚组患者临床症状较轻,部分患者可仅表现为眼外肌受累,很少出现肌无力危象;也有研究发现,LRP4抗体阳性患者均为GMG,表现为严重的肢带肌无力和/或进行性延髓麻痹。目前研究尚未发现LRP4-MG伴有胸腺异常。 1.3.5 抗体阴性MG:极少部分患者血清无上述可检测到的抗体,包括AChR、MuSK及LRP4抗体,称为抗体阴性MG。 1.3.6 胸腺瘤相关MG:约占MG患者的10%-15%,属于副肿瘤综合征,任何年龄均可发病,相对发病高峰在50岁左右。绝大多数胸腺瘤相关MG可检测出AChR抗体,除此之外,多合并连接素(Titin)抗体及RyR抗体,胸腺瘤相关MG病情略重,需要更长疗程免疫抑制治疗。 二、MG辅助检查 2.1 药理学检查 甲硫酸新斯的明试验:成人肌肉注射1.0-1.5mg,同时予以阿托品0.5mg肌肉注射,以消除其M胆碱样不良反应;儿童可按体重0.02-0.04mg/kg,最大用药剂量不超1.0mg。注射前可参照MG临床绝对评分标准,选取肌无力症状最明显的肌群,记录1次肌力,注射后每10min记录1次,持续记录60min。以改善最显著时的单项绝对分数,按照下列公式计算相对评分作为试验结果判定值。相对评分=(试验前该项记录评分-注射后每次记录评分)/试验前该项记录评分×100%。相对评分≤25%为阴性,25%-60%为可疑阳性,≥60%为阳性。 2.2 电生理检查2.2.1 RNS:采用低频(2-3Hz)重复电刺激神经干,在相应肌肉记录复合肌肉动作电位(compound muscle action potentials,CMAP)。常规检测的神经包括面神经、副神经、腋神经和尺神经。持续时间为3s,结果以第4或第5波与第1波的波幅比值进行判断,波幅衰减10%以上为阳性,称为波幅递减。部分患者第4波后波幅不再降低和回升,形成U字样改变。服用胆碱酯酶抑制剂的患者需停药12-18h后进行检查,但需充分考虑病情。与突触前膜病变鉴别时需要进行高频RNS(30-50Hz)或者大力收缩后10s观察CMAP波幅变化,递增100%以上为异常,称为波幅递增。 2.2.2 单纤维肌电图(SFEMG):使用特殊的单纤维针电极测量同一神经肌纤维电位间的间隔是否延长来反映NMJ处的功能,通过测定“颤抖”(Jitter)研究神经-肌肉传递功能。“颤抖”一般为15-35μs,超过55μs为“颤抖增宽”,一块肌肉记录20个“颤抖”中有2个或2个以上大于55μs则为异常。检测过程中出现阻滞(block)也判定为异常。SFEMG并非常规的检测手段,敏感性高。SFEMG不受胆碱酯酶抑制剂影响,主要用于OMG或临床怀疑MG但RNS未见异常的患者。 2.3 血清抗体检测 2.3.1 抗AChR抗体:约50%-60%的OMG、85%-90%的GMG血清中可检测到AChR抗体。需注意的是AChR抗体检测结果为阴性时不能排除MG诊断。放射免疫沉淀法(radioimmunoprecipitation assay,RIA)是AChR抗体的标准检测方法,可进行定量检测。ELISA法较RIA法敏感性低。 2.3.2 抗MuSK抗体:在10%-20%的AChR抗体阴性MG患者血清中可检测到MuSK抗体,标准检测方法为RIA或ELISA。 2.3.3 抗LRP4抗体:在7%-33%的AChR、MuSK抗体阴性MG患者中可检测出LRP4抗体。 2.3.4 抗横纹肌抗体:包括抗Titin和RyR抗体。Titin抗体通常采用ELISA法检测,RyR抗体可采用免疫印迹法或ELISA法检测。 2.4 胸腺影像学检查 约80%左右的MG患者伴有胸腺异常,包括胸腺增生及胸腺瘤。CT为常规检测胸腺方法,胸腺瘤检出率可达94%;MR有助于区分一些微小胸腺瘤和以软组织包块为表现的胸腺增生;必要时可行CT增强扫描;PET-CT有助于区别胸腺癌和胸腺瘤。 2.5 合并其他自身免疫性疾病检测 MG患者可合并其他自身免疫病,如自身免疫性甲状腺疾病,最常见的是Graves病,其次为桥本甲状腺炎。OMG合并自身免疫性甲状腺疾病比例更高,因此,MG患者需常规筛查甲状腺功能及甲状腺自身抗体、甲状腺超声检查观察有无弥漫性甲状腺肿大,以及其他自身免疫性疾病相关抗体检测。 三、MG诊断与鉴别诊断 3.1 诊断依据 在具有典型MG临床特征(波动性肌无力)的基础上,满足以下3点中的任意一点即可做出诊断,包括药理学检查、电生理学特征以及血清抗AChR等抗体检测。同时需排除其他疾病。所有确诊MG患者需进一步完善胸腺影像学检查(纵隔CT或MRI),进一步行亚组分类。 3.2 鉴别诊断 3.2.1 与OMG的鉴别诊断: (1)眼睑痉挛:发病年龄较大,表现为过度瞬目动作,可伴有眼部干燥、刺激感(需排除干燥综合征),可能会出现长时间闭眼,误认为是上睑下垂;强光刺激可加重眼睑痉挛,患者需长期戴墨镜;触摸眼角、咳嗽和说话时眼睑痉挛可得到意外改善。氟哌啶醇、阿立哌唑或者氯硝西泮治疗有效。 (2)Miller-Fisher综合征:属于Guillain-Barré综合征变异型,表现为急性眼外肌麻痹、共济失调和腱反射消失,也可表现为单纯的眼外肌麻痹型,易误诊为MG;肌电图检查示神经传导速度减慢,脑脊液检查可见蛋白-细胞分离现象,部分患者血清可检测出抗GQ1b抗体或GT1a抗体。 (3)慢性进行性眼外肌麻痹(chronic progressive external ophthalmoplegia,CPEO)或Kearn-Sayre综合征(KSS):属于线粒体脑肌病,CPEO表现为双侧进展性无波动性眼睑下垂、眼外肌麻痹,可伴近端肢体无力。若同时合并视网膜色素变性、小脑萎缩以及心脏传导阻滞,即为KSS综合征。肌电图检查示肌源性损害,少数患者可伴有周围神经传导速度减慢。血乳酸轻度增高,肌肉活检和基因检查有助于确诊。 (4)眼咽型肌营养不良(oculopharyngeal muscular dystrophy):为常染色体显性遗传,存在家族史;表现为老年起病的无波动性对称性眼睑下垂,斜视明显,但无复视,逐渐出现吞咽困难、构音障碍。肌电图检查提示肌源性损害。血清肌酶多正常或轻度增高,肌肉活检和基因检测有助于诊断。 (5)脑干病变:包括脑干缺血性卒中、肿瘤、副肿瘤综合征、Wernicke脑病、视神经脊髓炎谱系疾病、Bickerstaff脑干脑炎及其他感染性脑炎,均可以急性双睑下垂为首发症状,易于与MG混淆,结合病史、头颅MRI以及特异性抗体检测有助于明确诊断。 (6)眶内占位病变:如眶内肿瘤、脓肿或炎性假瘤等,可表现为眼外肌麻痹并伴结膜充血、眼球突出、眼睑水肿。眼眶MRI、CT或超声检查有助于诊断。 (7)脑神经麻痹(III、IV、VI):一侧海绵窦感染、肿瘤、非特异性炎症、颈内动脉海绵窦瘘均可表现为单侧眼睑下垂、眼外肌麻痹伴疼痛,头颅MRI及脑脊液检查有助于鉴别诊断。此外,糖尿病也可引起单纯动眼神经或外展神经麻痹。 (8)Graves眼病:属于自身免疫性甲状腺疾病,表现为自限性眼外肌无力、眼睑退缩,不伴眼睑下垂。眼眶CT或MRI检查显示眼外肌肿胀,甲状腺功能亢进或减退,抗甲状腺球蛋白抗体、抗甲状腺微粒体抗体或抗促甲状腺激素受体抗体阳性。 (9)先天性肌无力综合征(congenital myasthenic syndromes,CMS):是一组罕见的由编码NMJ结构及功能蛋白的基因突变所致NMJ传递障碍的遗传性疾病,依据突变基因编码蛋白在NMJ的分布,CMS可分为突触前、突触以及突触后突变。CMS临床表现异质性很大,极易被误诊为抗体阴性的MG、线粒体肌病等。多在出生时、婴幼儿期出现眼睑下垂、睁眼困难、喂养困难及运动发育迟滞等症状。青春期逐渐出现眼球固定,与MG在临床及电生理表现类似,鉴别主要依靠血清学抗体检测及全外显子测序。 3.2.2 与GMG的鉴别诊断: (1)Lambert-Eaton肌无力综合征(LEMS):是免疫介导的累及NMJ突触前膜电压门控钙通道(voltage-gated calcium channel,VGCC)的疾病,属于神经系统副肿瘤综合征,多继发于小细胞肺癌,也可继发于其他神经内分泌肿瘤。临床表现:四肢近端对称性无力,腱反射减低,以口干为突出表现的自主神经症状,极少出现眼外肌受累,腱反射在运动后可短暂恢复,其他自主神经症状如便秘、性功能障碍、出汗异常较少见。RNS为低频刺激(2-3Hz)出现CMAP波幅递减大于10%;高频刺激(20-50Hz)或者大力收缩后10sCMAP波幅递增大于60%或100%。血清VGCC抗体多呈阳性,合并小细胞肺癌的LEMS可同时出现SOX-1抗体阳性。 (2)运动神经元病(进行性延髓麻痹):尤其需与MuSK-MG相鉴别,患者均以延髓症状为突出表现,进行性延髓麻痹可出现上运动神经元损害证据;若患者病程较长,病程中出现眼睑下垂及复视,缺乏上运动神经元损害的证据,需警惕有无MuSK-MG的可能,建议行MuSK抗体检测。 (3)CMS:CMS临床表现异质性大,DOK7、RAPSN、CHAT以及GFPT1突变所致CMS几乎不出现眼外肌麻痹。GFPT1突变所致CMS可表现为四肢肌易疲劳,肌活检可见管聚集或空泡样改变,GMPPB突变所致CMS血清肌酶明显升高,肌活检提示为肌营养不良样改变;CMS肌电图可表现为肌源性损害。因此,肌肉活检及高通量全外显子测序有助于确诊。 (4)肉毒中毒:由肉毒杆菌毒素累及NMJ突触前膜所致,表现为眼外肌麻痹以及吞咽、构音、咀嚼无力,肢体对称性弛缓性瘫痪,可累及呼吸肌。若为食物肉毒毒素中毒,在肌无力之前可出现严重恶心、呕吐。瞳孔扩大和对光反射迟钝、四肢腱反射消失、突出的自主神经症状有助于将肉毒中毒与MG鉴别。电生理检查结果与LEMS相似:低频RNS可见波幅递减,高频RNS波幅增高或无反应,取决于中毒程度。对血清、粪便及食物进行肉毒杆菌分离及毒素鉴定可明确诊断。 (5)Guillain-Barré综合征:为免疫介导的急性炎性脱髓鞘性周围神经病,表现为弛缓性肢体无力,感觉丧失、腱反射减低或消失。肌电图示运动感觉神经传导末端潜伏期延长,传导速度减慢,传导波幅降低;脑脊液检查可见蛋白-细胞分离现象。咽颈臂丛型Guillain-Barré综合征(PCB)以球麻痹、抬颈及双上肢近端无力为主要表现,易误诊为MG,尤其是MuSK-MG。PCB多有前驱感染病史,查体可见双上肢腱反射减低或消失,脑脊液可出现蛋白-细胞分离现象,血清抗GT1a抗体可呈阳性,与Fisher综合征共病时,GQ1b抗体也可呈阳性。 (6)慢性炎性脱髓鞘性多发性神经病:免疫介导的慢性运动感觉周围神经病,表现为弛缓性四肢无力,套式感觉减退,腱反射减低或消失。肌电图示运动、感觉神经传导速度减慢,波幅降低和传导阻滞。脑脊液可见蛋白-细胞分离现象,周围神经活检有助于诊断。 (7)炎性肌病:多种原因导致的骨骼肌间质性炎性病变,表现为进行性加重的弛缓性四肢无力和疼痛。肌电图示肌源性损害。血肌酶明显升高、肌肉活检有助于诊断。糖皮质激素治疗有效。 (8)代谢性肌病:如肌肉代谢酶、脂质代谢或线粒体受损所致肌肉疾病表现为弛缓性四肢无力,不能耐受疲劳,腱反射减低或消失,伴有其他器官损害。肌电图示肌源性损害。血肌酶正常或轻微升高。肌活检及基因检测有助于诊断。

韩小磊
主治医师
河南省中医院
164
人阅读
查看详情
文章 左旋多巴反应实验
具体步骤: (1) 医生或者研究助手预约时间段,利用MDS国际公认的帕金森病统一评估量表的III部分(MDS-UPDRSIII)来评估患者停药后的运动症状。空腹状态下,先进行UPDRS-Ⅲ评分作为基线。 (2) 评估完毕之后,让患者口服吗丁啉10 mg(避免大剂量药物引起胃肠道反应如呕吐等),30分钟后一次性嚼服换算的美多芭总剂量,注意:美多芭总剂量=患者清晨所服药物折算成美多芭的总剂量*150%),此后每隔30分钟进行1次UPDRS-Ⅲ评分,至服药后4h,观察到症状逐渐改善又逐渐加重这一过程后结束评估,整个过程约需4小时左右。 (3) 在此过程中,建议对评估过程进行录像,特别是对患者最差的状态(服药前"关”的状态)和最好的状态(服药后完全“开”的状态)的录像。 (4) 计算UPDRS的最大改善率,最大改善率=(服药前基线评分-服药后最低评分)/服药前基线评分×100%。 注意事项:(1) 暂时停用所有抗帕金森病药物,多巴胺受体激动剂(例如普拉克索、吡呗地尔、罗替高汀、罗匹尼罗等)停用72小时,多巴制剂(美多巴、达灵复、息宁)停用12小时后方可进行该试验。 (2) 尤其是多巴制剂,患者如果预约时间是上午8:00,那前一天晚上20:00以后就不能再服药多巴制剂,否则影响评估的准确性,这一点非常重要。 (3) 如果患者药效减退的时间段非常难受,无法坚持停药12小时,可以根据患者的病情允许患者改在“关期”评估。 (4) 评估可以看情况安排在门诊或者病房进行,患者要带上平时的药物、早餐、水,并在家人陪同下来医院进行评估。 药物换算表(介绍3个表,任选其中一个即可):
韩小磊
主治医师
河南省中医院
172
人阅读
查看详情
文章 指南共识 | 中国重症肌无力诊断和治疗指南(二、治疗)
MG治疗 4.1 治疗目标及相关定义 4.1.1 治疗目标:依据MGFA对MG干预后状态(post-intervention status)的分级(表4),达到微小状态(minimal manifestation status,MMS)或更好,治疗相关副作用(common terminology criteria for adverse events,CTCAE)≤1级。 4.1.2 相关定义:(1)MMS:没有任何因肌无力引起的功能受限,经专业的神经肌病医生检查可发现某些肌肉无力。(2)CTCAE1级:该治疗未引起临床症状或症状轻微,不需要干预。(3)危象前状态(impending myasthenic crisis):MG病情快速恶化,依据临床医生的经验判断,数天或数周内可能发生肌无力危象(manifest myasthenic crisis)。危象前状态的及时识别、干预可避免肌无力危象的发生。(4)肌无力危象:MG病情快速恶化,需要立即开放气道,辅助通气;或者MGFA分型为V型。(5)难治性MG(refractory MG):对于难治性MG尚无统一的标准,基于现有研究证据定义为:传统的糖皮质激素或者至少2种免疫抑制剂(足量、足疗程)治疗无效,干预后状态为无变化或者加重;不能耐受免疫抑制剂的副作用或有免疫抑制剂使用禁忌证,需要反复给予IVIG或者PE以缓解病情;或病程中反复出现肌无力危象。 4.2 急性加重期治疗 IVIG与PE主要用于病情快速进展、危及生命的情况,如肌无力危象、严重的球麻痹所致吞咽困难、肌无力患者胸腺切除术前和围手术期治疗,可使绝大部分患者的病情得到快速缓解。为达到持续缓解,可同时启动免疫抑制治疗(非激素类免疫抑制剂),因激素早期可一过性加重病情,甚至诱发肌无力危象,于IVIG与PE使用后症状稳定时添加激素治疗。IVIG多于使用后5-10d左右起效,作用可持续2个月左右。在稳定的中、重度MG患者中重复使用并不能增加疗效或减少糖皮质激素的用量。(1)IVIG使用方法:按体重400mg/(kg·d)静脉注射5d。副作用包括头痛、无菌性脑膜炎、流感样症状和肾功能损害等,伴有肾功能损害的患者禁用。(2)PE使用方法:剂量为1.0-1.5倍总血浆容量,在10-14d内进行3-6次置换,置换液可用健康人血浆或白蛋白。多于首次或第2次PE后2d左右起效,作用可持续1-2个月。副作用包括血钙降低、低血压、继发性感染和出血等。伴有感染的患者慎用PE,宜在感染控制后使用。如PE期间发生感染则要积极控制感染,并根据病情决定是否继续进行PE。 IVIG与PE在严重MG中的疗效相当,但需注意的是使用IVIG治疗后4周内不建议进行PE,这可能影响IVIG的效果。IVIG在轻型MG或OMG患者中的疗效不确定,对于MuSK-MG,推荐使用PE。此外,IVIG还可用于难治性MG或者免疫抑制剂治疗有禁忌的MG患者。 4.3 药物治疗4.3.1 胆碱酯酶抑制剂——症状性治疗:最常用的是溴吡斯的明,其是治疗所有类型MG的一线药物,可缓解、改善绝大部分MG患者的临床症状。溴吡斯的明应当作为MG患者初始治疗的首选药物,依据病情与激素及其他非激素类免疫抑制联合使用。用法:一般成年人服用溴吡斯的明的首次剂量为60mg(儿童根据具体年龄使用),口服,3-4次/d,全天最大剂量不超过480mg。应根据MG患者对溴吡斯的明的敏感程度进行溴吡斯的明剂量的个体化应用,达到治疗目标时可逐渐减量或停药。溴吡斯的明的副作用包括恶心、流涎、腹痛、腹泻、心动过缓及出汗增多等。妊娠期使用溴吡斯的明是安全有效的。 4.3.2 免疫抑制治疗:免疫抑制药物包括糖皮质激素和其他口服非激素类免疫抑制剂,如硫唑嘌呤(azathioprine,AZA)、他克莫司(tacrolimus,FK-506)、吗替麦考酚酯(mycophenolate mofetil,MMF)、环孢素、甲氨蝶呤(methotrexate)及环磷酰胺(cyclophosphamide)。非激素类免疫抑制剂在糖皮质激素减量以及预防MG复发中发挥重要作用。值得注意的是:目前尚无临床研究比较不同非激素类免疫抑制剂的疗效,因此,药物选择尚无统一标准,更多依赖于临床医生的经验。 (1)糖皮质激素:目前仍为治疗MG的一线药物,可使70%-80%的患者症状得到明显改善。主要为口服醋酸泼尼松以及甲泼尼龙。醋酸泼尼松按体重0.5-1.0mg/(kg·d)清晨顿服,最大剂量不超过100mg/d(糖皮质激素剂量换算关系为:5mg醋酸泼尼松=4mg甲泼尼龙),一般2周内起效,6-8周效果最为显著。75%轻-中度MG对200mg泼尼松具有很好反应,以20mg起始,每5-7d递增10mg,至目标剂量。达到治疗目标后,维持6-8周后逐渐减量,每2-4周减5-10mg,至20mg后每4-8周减5mg,酌情隔日口服最低有效剂量,过快减量可致病情复发。 为避免口服大剂量激素,治疗初期与其他非激素类口服免疫抑制剂联用,可更快达到治疗目标。使用糖皮质激素期间必须严密观察病情变化,约40%-50%的患者在服药2-3周内症状一过性加重并有可能诱发肌无力危象,尤其是晚发型、病情严重或球部症状明显的患者,使用糖皮质激素早期更容易出现症状加重,因此,对上述患者应慎用糖皮质激素,可先使用IVIG或PE使病情稳定后再使用糖皮质激素,并做好开放气道的准备。长期服用糖皮质激素可引起食量增加、体重增加、向心性肥胖、血压升高、血糖升高、白内障、青光眼、内分泌功能紊乱、精神障碍、骨质疏松、股骨头坏死、消化道症状等,应引起高度重视。及时补充钙剂和双磷酸盐类药物可预防或减轻骨质疏松,使用抑酸类药物可预防胃肠道并发症。 (2)AZA:与糖皮质激素联合使用,有助于激素减量以及防止疾病复发,作为GMG及部分OMG的一线用药。AZA起效较慢,多于服药后3-6个月起效,1-2年后可达全效,可使70%-90%的MG患者症状得到明显改善。使用方法:从小剂量开始,50mg/d,每隔2-4周增加50mg,至有效治疗剂量为止[儿童按体重1-2mg/(kg·d),成人2-3mg/(kg·d),分2-3次口服]。如无严重或/和不可耐受的不良反应,可长期服用。主要副作用包括骨髓抑制(白细胞减少、贫血、血小板减少)、肝功损害、脱发、流感样症状及消化道症状等,多发生在启动治疗的6周左右。硫代嘌呤甲基转移酶(thiopurine methyltransferase)表型或基因型检测可预测服用AZA过程中白细胞减少的风险。长期服用AZA,应密切监测血常规和肝肾功能,服药第1个月,每周监测血常规及肝肾功能;服药后前6个月,应每个月监测血常规及肝肾功能;此后每3个月监测血常规及肝肾功能。若白细胞计数低于4.0×10^9/L,应将AZA减量;若白细胞计数低于3.0×10^9/L或肝功能检测指标为正常值上限的3倍,应立即停药。 (3)他克莫司:与环孢素作用机制相似,通过抑制钙神经素发挥免疫调节作用,耐受性较好,肾毒性小。他克莫司适用于不能耐受激素和其他免疫抑制剂副作用或对其疗效差的MG患者,特别是RyR抗体阳性者。他克莫司起效快,一般2周左右起效,疗效呈剂量依赖性。使用方法:3.0mg/d,分2次空腹口服,或按体重0.05-0.10mg/(kg·d)。建议:可于服药或者调整药物剂量3-4d后筛查血药浓度,理想谷浓度为2-9ng/mL。研究表明,他克莫司谷浓度≥4.8ng/mL,92%的患者可达到MMS或更好状态。主要副作用包括血糖升高、血镁降低、震颤、肝肾功损害以及罕见的骨髓抑制。 (4)MMF:作用机制同AZA,更安全,耐受性好,长期使用可使大多数患者达到MMS或更好状态。使用方法:起始剂量0.5-1.0g/d,分2次口服;维持剂量1.0-1.5g/d,症状稳定后每年减量不超过500mg/d,突然停药或快速减量可导致病情复发及恶化。MMF不可与AZA同时使用。常见不良反应为恶心、呕吐、腹泻、腹痛等胃肠道反应,白细胞减低,泌尿系统感染及病毒感染等。用药后的前6个月,每个月监测血常规及肝肾功,此后每3个月监测血常规及肝肾功能。MMF具有致畸性,备孕或怀孕妇女禁用。 (5)环孢素:通过干扰钙调神经磷酸酶信号,抑制包括白细胞介素2(IL-2)和γ干扰素在内的促炎细胞因子分泌,从而发挥免疫抑制作用。3-6个月起效,用于对激素及AZA疗效差或不能耐受其副作用的患者。环孢素早期与激素联合使用,可显著改善肌无力症状,并降低血中AChR抗体滴度,但肾毒性较大。使用方法:按体重2-4mg/(kg·d)口服,使用过程中应监测血浆环孢素药物浓度,推荐血药浓度为100-150ng/mL,并根据浓度调整环孢素剂量。主要副作用包括肾功损害、血压升高、震颤、牙龈增生、肌痛和流感样症状等。服药期间至少每个月监测血常规、肝肾功能1次,严密监测血压。因环孢素肾毒性较大以及和其他药物之间存在相互作用,不作为首选推荐。 (6)环磷酰胺:用于其他免疫抑制剂治疗无效的难治性及伴胸腺瘤的MG。与激素联合使用可显著改善肌无力症状,并在6-12个月时使激素用量减少。使用方法:成人静脉滴注400-800mg/周,或分2次口服,100mg/d,直至总量10-20g,个别患者需要服用到30g;儿童按体重3-5mg/(kg·d)分2次口服(不大于100mg),好转后减量,2mg/(kg·d)。儿童应慎用。副作用包括白细胞减少、脱发、恶心、呕吐、腹泻、出血性膀胱炎、骨髓抑制、致畸以及远期肿瘤风险等。每次使用前均需要复查血常规和肝肾功能。 (7)甲氨蝶呤:作为三线用药,用于其他免疫抑制剂治疗无效的难治性或伴胸腺瘤的MG。使用用法:口服,每周10mg起始,逐步加量至20mg/周,如不能耐受口服制剂产生的消化道不良反应,也可选择肌肉注射制剂,一般肌肉注射可使患者耐受更高的剂量。副作用包括胃肠道反应及肝功能异常,可伴发口腔炎、皮疹、肺纤维化、白细胞减低。治疗时需同时添加叶酸1mg/d预防口腔炎,并应密切关注骨髓抑制及肝功损害等副作用。甲氨蝶呤有生殖致畸性,怀孕或备孕妇女禁用。 4.4 靶向生物制剂 目前临床上用于MG治疗的靶向生物制剂包括已经被美国食品和药物监督管理局(FDA)批准使用的靶向补体的依库珠单抗(eculizumab),以及适应证外用药的靶向B细胞的利妥昔单抗(rituximab,RTX)。此外,一些靶向免疫系统不同组分的生物制剂仍在临床前研究,如靶向B细胞激活因子(B lymphocyte stimulating factor,BLyS)的Belimumab以及靶向FcRn的Efgartigimod等。 4.4.1 靶向B细胞治疗:RTX为人鼠嵌合的单克隆抗体,通过靶向B细胞膜分子CD20实现特异性清除B细胞,用于对激素和免疫抑制剂疗效差的难治性GMG,特别是MuSK-MG,对部分AChR-MG有效。RTX用药方案目前尚无统一标准,通常为诱导治疗序贯维持治疗。临床推荐诱导方案包括标准方案及低剂量方案。(1)标准方案:诱导剂量按体表面积375mg/m2,间隔1周给药1次,连续给药4周,序贯给药1g,间隔2周治疗1次 ,共2次;(2)低剂量方案包括:按体表面积375mg/m2,间隔2周给药1次,共2次或100+500mg单次治疗。维持剂量为按体表面积375-750mg/m2。通常在给药后第4周,患者外周血B细胞比例可降至0,1次给药为1个循环,作用可维持6个月,6个月后B细胞开始爬升。维持治疗更多为经验性治疗,有医生建议临床复发时追加RTX治疗,也有医生建议每隔6个月给予一次RTX治疗。CD27+记忆B细胞的监测有助于判断疾病复发以及指导RTX追加给药。RTX主要副作用包括发热、寒战、支气管痉挛、白细胞减少、血小板减少和进行性多灶性白质脑病等。 4.4.2 补体抑制剂:补体在AChR-MG发病中发挥着重要作用。依库珠单抗为靶向补体级联反应的关键组分补体C5的人源化单克隆抗体,可有效抑制C5激活。一项关于依库珠单抗在MG有效性及安全性的III期临床研究(REGAIN identifier:NCT01997229)以及其开放性扩展研究显示:依库珠单抗对其他免疫抑制治疗无效的AChR抗体阳性GMG(AChR-GMG)有显著疗效,56%的患者可达到MMS或药物缓解。2017年FDA批准依库珠单抗用于AChR-GMG成年患者的治疗,其价格昂贵,建议用于中重度、难治性MG。Zilucoplan为另一类靶向补体C5的大环肽类新型抑制剂,可特异性结合C5,阻止C5裂解为C5a和C5b,同时可阻止C5b和C6的结合,双重作用可有效阻止补体级联反应。与依库珠单抗不同的是,Zilucoplan是一种可以自我给药的皮下注射制剂。研究表明(NCT03315130)Zilucoplan可使中重度AChR-GMG症状得到快速且持续的缓解。 4.4.3 其他生物制剂:(1)Belimumab:以BLyS为靶点的人源化IgG1-λ单克隆抗体,BLyS在B细胞激活、成熟及存活中发挥关键作用。Belimumab能够清除所有的浆细胞、激活B细胞及天然B细胞,但不能清除记忆B细胞。一项观察Belimumab在AChR-MG或MuSK-MG有效性的二期、随机双盲安慰剂对照研究(NCT01480596)结果显示,接受Belimumab治疗组在12-24周症状达到持续缓解的比例较安慰剂组更高。 (2)Efgartigimod(ARGX-113):靶向FcRn的抗体片段,其与FcRn的亲和力超过正常IgG抗体的Fc部分,Efgartigimod通过与FcRn结合阻断IgG循环,导致引起自身免疫疾病IgG抗体的快速消耗。Efgartigimod在MG治疗中的二期临床试验已经完成,与安慰剂比较,Efgartigimod可明显改善MG临床症状(NCT02965573);关键性III期临床试验(ADAPT)结果显示,67.7%接受Efgartigimod治疗的AChR-GMG患者达到治疗终点(NCT03669588)。 4.5 胸腺切除 4.5.1 伴胸腺瘤MG:合并胸腺瘤的MG应尽早行胸腺切除手术,经胸骨正中入路扩大胸腺切除已成为治疗胸腺瘤及合并胸腺增生MG的标准手术方式。扩大胸腺切除指的是在不损伤喉神经、左侧迷走神经及膈神经的前提下,安全切除肿瘤及异位的胸腺组织。异位胸腺组织大多数存在于前纵隔脂肪中,除此之外,还包括位于包膜、侧甲及横膈膜的脂肪组织。 4.5.2 非胸腺瘤OMG:对其他治疗无效的OMG患者可行胸腺切除,据报道缓解率为6%-50%。一项研究回顾性分析了110例行胸腺切除的OMG患者,中位随访33.5个月,84.6%的患者达到了完全缓解;一项荟萃分析显示,非胸腺瘤OMG可从胸腺切除获益,该疗效需多中心随机对照研究进一步证实。 4.5.3 非胸腺瘤GMG:针对非胸腺瘤AChR-GMG,推荐在疾病早期行胸腺切除,可减少其他免疫抑制剂使用。一项首个全球多中心随机对照研究(MGTX)发现,胸腺切除可长期改善AChR-GMG的临床症状,有助于激素减量和减少合并使用AZA等免疫抑制剂。MuSK-MG不推荐行胸腺切除。胸腺切除起效时间为6-24个月不等。部分MG患者经胸腺切除后可完全治愈,也有部分MG患者胸腺切除仍需长期免疫抑制治疗。 胸腺切除方式包括经典的经胸骨正中胸腺切除以及近年来广泛应用的微创手术切除胸腺,如电视辅助胸腔镜(video-assisted thoracoscopic surgery,VATS)及“达芬奇”系统机器人。微创手术已成为胸腺切除的主流术式,与开胸手术相比,微创手术创伤小,住院时间短,止痛药物使用少,创口外观处理效果更美观。目前尚无这两种术式的随机对照比较研究。胸腺切除需在患者病情相对稳定,能够耐受手术的情况下进行。若症状严重,除非怀疑高度恶性胸腺瘤者外,可先给予相应治疗,待病情稳定后再行手术,有助于减少、防止术后肌无力危象的发生。 4.6 自体造血干细胞移植(autologous hematopoietic stem cell transplant,AHSCT) AHSCT在MG中的研究仅为小样本病例报道。国内有学者使用体外纯化的自体外周血CD34+细胞移植治疗5例难治性MG,结果显示患者远期疗效好,耐受性良好。一项单中心研究对7例行AHSCT治疗的难治性MG进行长达12年随访,所有患者均不需要服用任何药物,达到完全缓解。AHSCT有望成为MG治疗的重要手段之一,尤其是难治、复发MG患者。 4.7 其他治疗 对于眼睑下垂者,可采用眼睑支架或胶带(eyelid crutches/tape),或通过手术来改善。眼肌手术对长期固定性斜视可能有效。 4.8 不同类型MG患者的治疗 4.8.1 儿童及JMG:中国JMG以眼肌型多见,并可自发缓解。因此,JMG以溴吡斯的明治疗为主,不能达到治疗目标时可添加激素及其他非激素类口服免疫抑制剂。激素具有抑制生长发育的副作用,应避免长期使用,若需要长期使用,必须采用最低有效剂量维持以减少不良反应。小剂量糖皮质激素(按体重0.25mg/kg)可有效缓解临床症状,且无相关治疗副作用。JMG可定期应用PE或者IVIG,作为免疫抑制剂的替代选择。胸腺切除在JMG治疗中证据不足,不作为常规推荐。 4.8.2 MG合并妊娠:(1)计划妊娠:如计划妊娠,应避免使用甲氨蝶呤和MMF等有致畸性的药物,若正在使用上述药物时,建议停药后方可妊娠。(2)孕期:MG患者怀孕后对症状有何影响目前尚无明确定论。多数患者的病情不会加重,也不会影响分娩的时间和方式。溴吡斯的明仍为妊娠期的一线用药,不推荐静脉使用胆碱酯酶抑制剂,可诱发子宫收缩;激素相对安全,可以服用;尽管研究证实AZA相对安全,但也有一小部分专家不推荐妊娠期使用AZA。妊娠子痫不推荐使用硫酸镁,因其可阻断NMJ,推荐使用巴比妥类药物。(3)分娩:提倡自然分娩;肌无力母亲分娩的新生儿可出现短暂性肌无力,应严密观察,一旦发生立即转移至新生儿监护室。 4.8.3 成人OMG:成人OMG,尤其是晚发型、合并胸腺瘤、AChR抗体阳性及RNS异常的患者,推荐早期使用激素及免疫抑制剂。尽管目前尚无随机对照研究的证据,但多项回顾性研究及荟萃分析结果表明,早期使用泼尼松及其他免疫抑制剂不仅可改善眼肌无力症状,还可防止OMG继发全身化。 4.8.4 成人GMG:激素和免疫抑制剂联合使用为成人GMG的一线治疗。伴有胸腺异常,如胸腺瘤或胸腺增生,应早期行胸腺切除。 4.8.5 难治性MG:可使用RTX、依库珠单抗或者大剂量环磷酰胺治疗,也可尝试胸腺切除及AHSCT。 4.8.6 MuSK-MG:MuSK-MG与AChR-MG在发病机制和临床表现均不同,MuSK-MG对激素反应好,急性期PE可迅速缓解肌无力症状,多项回顾性研究证实RTX可显著改善MuSK-MG的临床症状,延长复发时间以及降低激素用量。MuSK-MG不推荐胸腺切除。 4.8.7 危象前状态或肌无力危象:患者一旦确诊为危象前状态或肌无力危象,应积极给予快速起效治疗(IVIG或PE),同时评估其呼吸功能,监测动脉血气,并进一步判断肌无力危象的类型(表5)。 一旦出现呼吸衰竭(I型或II型),应及时气管插管,正压通气。筛查危象诱因,如是否由感染、手术或使用加重肌无力的药物所致,并积极采取相应控制措施(如控制感染、停用加重病情的药物等)。若为肌无力危象,酌情增加胆碱酯酶抑制剂剂量,直到安全剂量范围内(全天量小于480mg)肌无力症状改善满意为止,不主张静脉给予胆碱酯酶抑制剂,可增加呼吸道分泌物,导致气道管理困难;若为胆碱能危象,应停用胆碱酯酶抑制剂,酌情使用阿托品,一般5-7d后再次使用,从小剂量开始逐渐加量,目前胆碱能危象已很少见。机械通气的患者需加强气道护理,定时翻身、拍背、吸痰及雾化,积极控制肺部感染,逐步调整呼吸机模式,尽早脱离呼吸机。 4.8.8 ICIs相关MG(ICIs-MG):在使用ICIs治疗肿瘤的同时,引起既往MG病情加重或复发,以及ICIs治疗后新发的MG,可以同时合并肌炎及心肌炎。ICIs主要通过激活并促进T细胞抗肿瘤免疫,从而杀伤肿瘤细胞。ICIs包括细胞毒T淋巴细胞抗原4(CTLA-4)抑制剂(ipilimumab等)、程序性死亡受体1(PD-1)及其程序性死亡配体1(PD-L1)抑制剂(nivolumab、pembrolizumab等)。ICIs-MG病情较重,肌无力危象发生率高。需要更积极治疗,推荐大剂量甲强龙冲击联合IVIG或PE,是否需要停用ICIs需根据肿瘤治疗情况。 4.9 MG患者合并其他疾病 MG患者可合并Graves病、多发性肌炎、多发性硬化、干燥综合征、周期性麻痹、Hashimoto病、类风湿性关节炎、系统性红斑狼疮、Guillain-Barré综合征、再生障碍性贫血等疾病,部分患者还可能累及心肌,表现为心电图异常、心律失常等。因此,在积极治疗MG的同时,还要兼顾可能合并的其他疾病。 4.10 治疗MG过程中需注意的事项 MG患者慎用的药物包括:部分激素类药物、部分抗感染药物(如氨基糖甙类抗生素等以及两性霉素等抗真菌药物)、部分心血管药物(如利多卡因、奎尼丁、β-受体阻滞剂、维拉帕米等)、部分抗癫痫药物(如苯妥英钠、乙琥胺等)、部分抗精神病药物(如氯丙嗪、碳酸锂、地西泮、氯硝西泮等)、部分麻醉药物(如吗啡、哌替啶等)、部分抗风湿药物(如青霉胺、氯喹等)。其他注意事项包括:禁用肥皂水灌肠;注意休息、保暖;避免劳累、受凉、感冒、情绪波动等。 随着免疫抑制治疗在MG的广泛应用,绝大部分患者预后得到了明显改善,肌无力危象发生率和死亡率明显降低。许多靶向免疫系统不同组分的生物制剂,如靶向B细胞和补体的单克隆抗体,使少数难治性MG的病情得到有效控制。胸腺切除在非胸腺瘤AChR-GMG治疗中获得了更多研究证据的支持,将成为此类患者的治疗选择之一。早期免疫抑制治疗可防止OMG继发全身化,有望成为高转化风险OMG的标准化治疗。总之,MG临床表现具有很大异质性,在临床实践中,需考虑患者的发病年龄、疾病严重程度、是否合并胸腺瘤、血清学特点、治疗并发症以及治疗费用等,尽量做到安全、有效、精准化治疗。
韩小磊
主治医师
河南省中医院
152
人阅读
查看详情
相关问诊
失眠,经常难以入睡,睡眠质量差,伴有心理恐惧。患者女性21岁
就诊科室:中医神经内科
总交流次数:21
医生建议:失眠可能与心理因素有关,建议调整生活习惯,改善睡眠环境,必要时可考虑使用安眠药物。生活上,保持规律作息,避免过度紧张,适当锻炼身体,有助于改善睡眠质量。
韩小磊
主治医师
河南省中医院
查看详情
患者头晕,已做脑部CT,咨询关于磁共振检查及病情治疗建议。患者女性66岁
就诊科室:中医神经内科
总交流次数:55
医生建议:根据症状和检查结果,患者可能患有轻微脑梗塞。建议进行磁共振检查以明确诊断。治疗上以药物和点滴治疗为主,需根据颅内血管情况判断是否需要手术。生活中注意保持健康的生活方式,避免过度劳累和情绪波动。如有新发脑梗塞,需及时治疗并遵医嘱进行用药。
韩小磊
主治医师
河南省中医院
查看详情
开车时犯恶心,非孕期,无生理期,询问缓解方法。患者女性15岁
就诊科室:中医神经内科
总交流次数:8
医生建议:孕期恶心可能是由于紧张或胃部不适引起,建议避免空腹、少食多餐,可尝试按内关穴缓解。如症状严重,请咨询医生是否需要用药。生活上,建议进行轻松活动,如散步,并保持良好的饮食习惯。
韩小磊
主治医师
河南省中医院
查看详情
