糖皮质激素类药物的常见不良反应及注意事项

全球医疗视野
关注全球医疗动态,介绍不同国家和地区的医疗制度、技术和经验
醋酸氢化可的松片是一种常用的糖皮质激素类药物,主要用于治疗肾上腺皮质功能减退症和先天性肾上腺皮质增生症。在应用生理剂量替代治疗时,糖皮质激素一般无明显不良反应。然而,不良反应多发生在应用药理剂量时,且与疗程、剂量、用药种类、用法及给药途径等因素密切相关。
甾体激素类药物常见的不良反应包括:
- 长程使用可能引起医源性库欣综合征面容和体态、体重增加、下肢浮肿、紫纹、易出血倾向、创口愈合不良、痤疮、月经紊乱、肱或股骨头缺血性坏死、骨质疏松及骨折、肌无力、肌萎缩、低血钾综合征、胃肠道刺激、胰腺炎、消化性溃疡或穿孔、儿童生长受到抑制、青光眼、白内障、良性颅内压升高综合征、糖耐量减退和糖尿病加重。
- 患者可能出现精神症状,如欣快感、激动、谵妄、不安、定向力障碍,也可表现为抑制。
- 并发感染为肾上腺皮质激素的主要不良反应,包括真菌、结核菌、葡萄球菌、变形杆菌、绿脓杆菌和各种疱疹病毒。
- 糖皮质激素停药综合征,有时患者在停药后出现头晕、昏厥倾向、腹痛或背痛、低热、食欲减退、恶心、呕吐、肌肉或关节疼痛、头疼、乏力、软弱等症状。
醋酸氢化可的松片禁用于对本品及其它甾体激素过敏者,以及患有严重的精神病、活动性消化性溃疡病、新近胃肠吻合手术、骨折、创伤修复期、角膜溃疡、肾上腺皮质机能亢进症、高血压、糖尿病、孕妇、抗菌药物不能控制的感染等疾病的患者。
在使用醋酸氢化可的松片时,需要注意以下几点:
- 诱发感染:在激素作用下,原来已被控制的感染可活动起来,最常见者为结核感染复发。在某些感染时应用激素可减轻组织的破坏、减少渗出、减轻感染中毒症状,但必须同时用有效的抗生素治疗、密切观察病情变化,在短期用药后,即应迅速减量、停药。
- 对诊断的干扰:糖皮质激素可使血糖、血胆固醇和血脂肪酸、血钠水平升高、使血钙、血钾下降。对外周血象的影响为淋巴细胞、真核细胞及嗜酸、嗜碱细胞数下降,多核白细胞和血小板增加,后者也可下降。长期大剂量服用糖皮质激素可使皮肤试验结果呈假阴性,如结核菌素试验、组织胞浆菌素试验和过敏反应皮试等。
- 慎用情况:心脏病或急性心力衰竭、糖尿病、憩室炎、情绪不稳定和有精神病倾向、全身性真菌感染、青光眼、肝功能损害、眼单纯性疱疹、高脂蛋白血症、高血压、甲减、重症肌无力、骨质疏松、胃溃疡、胃炎或食管炎、肾功能损害或结石、结核病等。
- 随访检查:长期应用糖皮质激素者,应定期检查血糖、尿糖或糖耐量试验、小儿生长和发育情况、眼科检查、血清电解质和大便隐血、高血压和骨质疏松的检查。

推荐医生列表

李莉
长治医学院附属和济医院内分泌科
好评100%|接诊量381|响应时长4分钟
擅长:擅长糖尿病及其并发症、甲状腺疾病、骨质疏松症、血脂异常、痛风及高尿酸血症、垂体疾病、肾上腺占位及其引起的内分泌性高血压、先天性肾上腺皮质增生症、儿童及青春期生长发育、Bartter综合征等疾病的诊治。
¥20.0起
问医生

孟凡东
徐州医科大学附属医院内分泌科
好评100%|接诊量87|响应时长18分钟
擅长:对糖尿病、甲亢、甲状腺结节、胰岛素抵抗、身材矮小、低钾、内分泌性高血压等内分泌代谢系统常见病、多发病的诊治已日臻成熟;对内分泌科少见病如先天性肾上腺皮质增生症、特发性低促性腺激素性性腺功能减退、胰岛细胞瘤有独到见解;对内分泌急症如糖尿病乳酸酸中毒、甲亢危象、垂体危象等有一套较为完善的诊断和抢救措施。
¥30.0起
问医生
本文由作者上传,文章内容仅供参考。如有相关事宜可联系jdh-hezuo@jd.com

疾病科普
查看更多
本病为常见的隐形遗传病,女孩多见临床表现为性征发育异常、电解质代谢紊乱(低钾性碱中毒)等需长期药物治疗,预后取决于病情及治疗时间的选择简介先天性肾上腺皮质增生症是一种罕见遗传病,是由于肾上腺皮质激素合成中酶缺乏所致,患者临床表现取决于酶缺陷部位及缺陷严重程度。常见症状有呕吐、腹泻、女性男性化(阴唇、阴蒂如男孩的阴茎、阴囊)、男孩性早熟(未发育前出现阴毛、胡须)、高血压、身材矮小等,严重者可致新生儿死亡。通过患者临床表现,结合实验室检查17-酮类固醇、17-羟孕酮水平、染色体分析基因突变、影像学检查判断骨龄及肾上腺情况来确诊。目前主要通过药物治疗缓解患者症状,保证儿童正常生长发育,部分女性患者可行手术矫正性器官畸形。刚出生就开始治疗预后更好,可保证患儿正常生长,因此本病需早诊早治。症状表现:患者可有女性男性化(阴唇、阴蒂外形如男孩的阴茎、阴囊)、男孩性早熟(男孩早期出现阴毛、胡须、阴茎大于正常儿童)、呕吐、腹泻、骨龄提前、高血压、脱水(表现为头晕、皮肤弹性差、眼窝凹陷)、低钾性碱中毒(可表现为四肢麻木、精神萎靡、腹胀及便秘等)、男孩假两性畸形(男孩外生殖器女性化、乳房发育)及骨骼畸形。诊断依据:根据患者临床表现,结合实验室检查17-酮类固醇、17-羟孕酮水平、染色体分析基因突变、影像学检查判断骨龄及肾上腺情况来确诊。先天性肾上腺皮质增生症有哪些类型?根据酶缺陷类型同,可分为:21-羟化酶缺乏症(最常见,占本病90%~95%);11β羟化酶缺乏症;3β-羟类固醇脱氢酶缺乏症;17α-羟化酶缺乏症;类脂性先天性肾上腺皮质增生症;细胞色素P450氧化还原酶缺乏症[1]。是否具有传染性?是否常见?本病罕见,在新生儿中发病率为1/20000~1/16000[1]。是否可以治愈?法治愈。只能通过药物治疗控制症状,保证儿童正常生长发育,避免并发症。是否遗传?是是否医保范围?是
 王建伟副主任医师北京积水潭医院
王建伟副主任医师北京积水潭医院
大家还在看

龟头周围痒痛、红肿,可能是发炎了?
生物医疗创新站

先天性肾上腺皮质增生症会表现出哪些症状?
#儿童#疾病概述#全身症状

JDH健康大讲堂

早泄的治疗:达怕西丁和其他方法
数字健康领航者
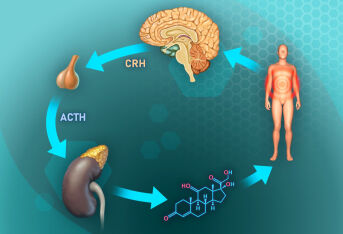
先天性肾上腺皮质增生症常用药物及用药期间注意事项有哪些?
#儿童#疾病概述#药物治疗
JDH健康大讲堂
肾上腺皮质增生是指什么?
#病因#中医科#常见症状#疾病人群#亚健康人群#健康养生人群

主治医师周喜玉

我们需要掌握一些常识避免延误病情
#产后护理#概述#消化内科#疾病人群#亚健康人群#孕婴童人群

副主任医师王明轩
肾上腺皮质增生症有什么症状?
#中医科#常见症状#肾病内科#一般治疗#疾病人群#亚健康人群#健康养生人群
主治医师周喜玉

包皮手术后第三天,阴茎疼痛正常吗?
生命之光传递者
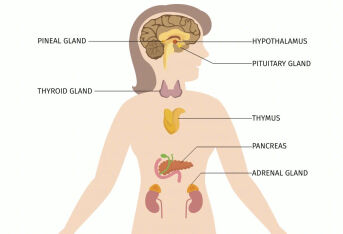
先天性肾上腺皮质增生症是什么?
#疾病概述#全身症状
JDH健康大讲堂

儿童先天性肾上腺皮质增生症
#儿童#检查#病因#小儿症状#一般治疗

住院医师陈晓春

包皮过长引起的尿道炎症怎么办?
医者荣耀

21岁才翻开包皮,龟头太敏感怎么办?
智慧医疗先锋者

性激素六项检查
#检查#诊断#妇产科#疾病人群#亚健康人群#健康养生人群

主治医师阮仙梅
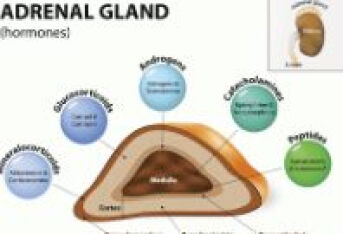
先天性肾上腺皮质增生症怎么办?
#运动健身#病因#诊断#中医科#疾病人群#亚健康人群#健康养生人群#孕婴童人群
主治医师周喜玉
*本站内容仅供医学知识科普使用,任何关于疾病、用药建议都不能替代执业医师当面诊断,请谨慎参阅