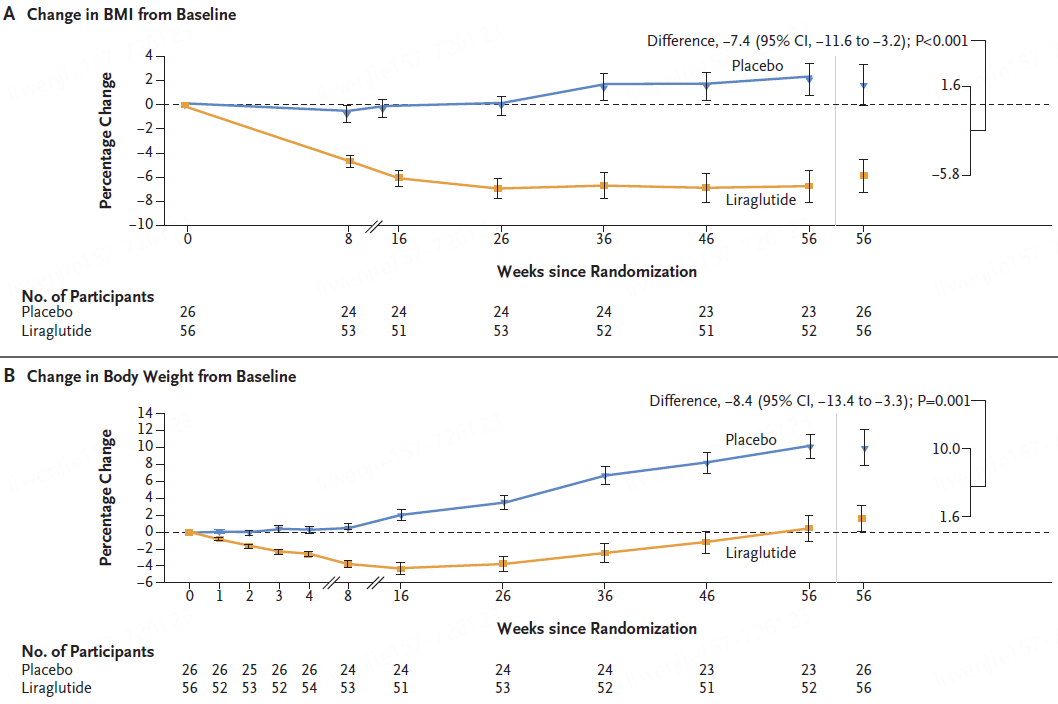
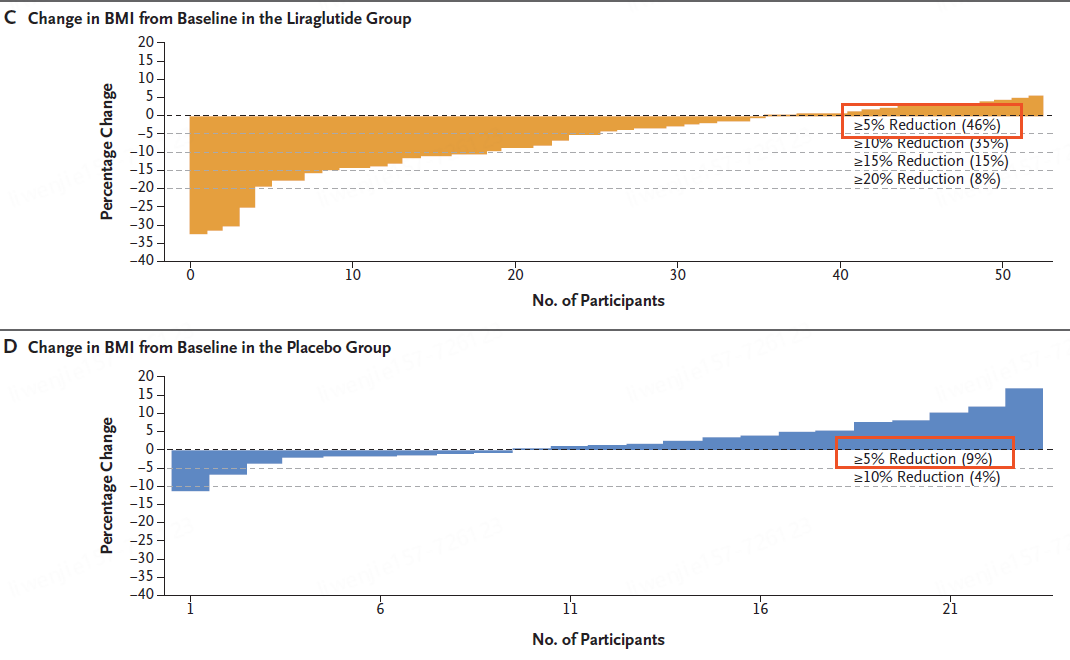
无锡市第九人民医院,暨无锡市骨科医院、苏大附属无锡九院,创建于1989年9月,是一所集医疗、保健、预防、急救、康复、教学、科研于一体的现代化综合性医院。作为江苏省首家三级甲等骨科医院,率先架构“八位一体”大骨科诊疗格局,现为国家卫健委手功能重建重点实验室手外科显微外科教育基地,中华医学会显微外科技术培训基地,中国医师协会显微外科技能培训中心,国家级住院医师规范化培训协同基地,江苏省工伤康复定点机构,中国创伤救治联盟创伤中心建设单位。设有顾玉东院士工作站,邱贵兴院士工作站,博士后科研工作站。医院交通便利,环境优美,坐落在风景秀丽的惠山森林公园旁,地铁二号线上。医院核定床位979张,现有卫技人员940名,其中高级职称147名,教授、副教授6名,博士生、硕士生导师21名,博士42名,硕士200名。享受国务院特殊津贴专家2名,江苏省医学领军人才、省卫生领军人才、省333工程培养对象、省突贡专家等21人,无锡市名医、市杰出医学人才、市医学领军人才等17人。医院开设手外科、创伤骨科、足踝外科、脊柱外科、关节外科、小儿骨科、运动医学科、创面修复科、康复医学科、内科、外科、妇产科、儿科、眼科、耳鼻咽喉头颈外科、重症医学科、急诊医学科、麻醉科、中医科等25个临床一级科室,以及心血管内科、神经内科、消化内科、肾内科、呼吸与危重症医学科、内分泌科、普通外科、肛肠科、泌尿外科、神经外科、胸外科、口腔科、皮肤科等21个临床二级科室,设有医学影像科、超声医学科、医学检验科、输血科、病理科、药学部、体检中心等8个医技一级科室。近五年来,共获得国家级、省、市科技成果奖16项,获得国家发明专利28项、实用新型专利322项,获得国家级、省、市科研立项资助133项。发表论文613篇,其中SCI收录133篇,北大中文核心期刊223篇。参与编著23本。举办国家级继续教育项目及研讨会36次,省、市级73次,培训学员7000余名。多年来,在各级政府及上级部门的正确领导和关心支持下,医院始终贯彻“科教兴院、人才强院、品牌办院”的发展理念,坚持“以病人为中心,以质量为核心,以服务献爱心”的服务宗旨,坚持公益属性,以建设人民满意医院为目标,全面推行精细化管理。医院先后获得“全国工人先锋号”“江苏省文明单位”“江苏省平安医院”“江苏省五一劳动奖状”“江苏省模范职工之家”“江苏省卫生健康行业先进基层党组织”“无锡市文明单位”等荣誉。先天性巨结肠又称希尔施普龙病。由于结肠缺乏神经节细胞导致肠管持续痉挛,粪便淤滞于近端结肠,近端结肠肥厚、扩张,是小儿常见的先天性肠道疾病之一。,先天性因素,结肠,1.保守治疗痉挛肠段短、便秘症状轻者 可暂采用综合性非手术疗法,包括定时用等渗盐水洗肠(灌洗出入量要求相等,忌用高渗、低渗盐水或肥皂水),扩肛、甘油栓、缓泻药,避免粪便在结肠内淤积。若以上方法治疗无效,虽为短段巨结肠亦应手术治疗。 2.结肠造瘘 保守治疗失败或患者病情严重、不具备接受根治手术条件患儿,均适用结肠造瘘术; 3.根治手术 主要手术方式包括: (1)Swenson手术切除整个受累部位并且将正常肠管吻合在近肛门水平。 (2)Soave手术直肠内膜整个拉出,将保留的受累直肠外层套入正常的肠道内。 (3)Duhamel手术在肛门水平通过钳夹将未受累肠端吻合到直肠。,无,无,体格检查、X线片检查等。,。
 0
0
 0
0
 0
0
 0
0
 0
0
 0
0
 0
0
 0
0
 0
0
 0
0