









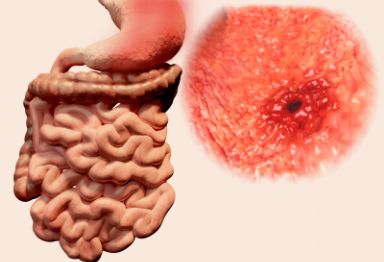
胃肠间质瘤(GISTs)是一种严重威胁人类健康的疾病,是起源于胃肠道间叶组织的肿瘤,占消化道间叶肿瘤的大部分,由于诊出率较低,长期被认为是一种罕见疾病。
近年来,随着诊疗水平的不断提高及临床研究的不断深入,其发病率也在不断上升,但遗憾的是仍然未能引起足够重视。主要是因其有一定的隐蔽性,在早期多无症状,有的呈球形生长,即便瘤体很大也不产生压迫感和出血。通常在体检或其他手术时偶尔发现。是名副其实的隐形杀手。
今天,我们就带您来认识一下这位“隐形的杀手”。
图片
一、胃肠间质瘤是不是癌症,如何确诊胃肠间质瘤?
我们通常所说的癌症是指起源上皮层的恶性肿瘤,由于间质瘤起源于间叶组织,所以并不是我们平时所说的癌症,而是具有一定恶性潜能的肿瘤,其恶性程度取决于肿瘤大小及有丝分裂指数来评估。
常规内镜、超声胃镜、CT 等可初步提示胃肠间质瘤,但是确诊需要穿刺标本或手术切除标本送检病理确诊。
二、胃肠间质瘤和其他的消化道肿瘤有什么区别?有什么特殊的症状?
作为一种胃肠道软组织肿瘤,胃肠间质瘤有别于“胃癌”或“肠癌”,可发生在消化道的任何部位。由于胃肠间质瘤常发于消化道内,长期被误认为是来源于平滑肌的肿瘤,故此被称为平滑肌瘤或平滑肌肉瘤。原因在于胃肠间质瘤不仅病程早期症状隐匿,其临床症状也无特异性可言,常常与胃肠道平滑肌瘤、胃肠道神经鞘瘤和胃肠道自主神经瘤等消化道肿瘤和疾病相混淆。
即使随着瘤体的增大,胃肠间质瘤会表现出一些明显症状,包括恶心、呕吐、腹痛、黑便和贫血等,但这些症状也多为非特异性,也可见于其他消化道疾病。这也导致胃肠间质瘤的准确诊断存在一定的难度。据目前文献报道在首次就诊的胃肠间质瘤患者中,有 20%~30% 的患者已发展成为晚期;有 11%~47% 的患者已发生肝转移和腹腔转移,错过了最佳治疗时机。所以体检筛查此病的必要性是显而易见的。
三、要筛查此类肿瘤,该做什么检查好呢?
检测间质瘤,常规胃肠镜、超声内镜、腹部 CT、磁共振(MRI)等都是可行的选择。但是由于症状缺乏特异性,胃肠间质瘤往往难以早期发现,尤其是直径小于 2 cm 的肿瘤可能没有任何症状,过半的间质瘤检出都是通过胃肠镜检查偶然发现的,由此可见常规体检中胃肠镜检查的重要性。
四、如果发现了胃肠道间质瘤,该怎么治疗?
对于临床上考虑为胃肠间质瘤的患者,应先进行临床评估,判定肿瘤部位、大小、是否局限、有无转移,综合评判进而决定治疗方式。
(1) 直径≤2 cm 的胃间质瘤伴临床症状者,可考虑行手术切除;无症状的拟诊间质瘤,应根据其内镜和内镜超声表现确定是否具有进展风险。内镜超声下的不良因素为边界不规整、溃疡、内部强回声和异质性,如合并不良因素,应考虑切除;如无不良因素,可定期进行内镜或影像学随访,时间间隔通常为 6~12 个月。对于难以接受反复的内镜检查,不能坚持随访者,可以考虑内镜下微创切除。
(2) 直径>2 cm 的胃间质瘤或其他部位的局限性胃肠间质瘤评估无手术禁忌证,建议根据位置及恶性潜能大小考虑内镜下微创切除或者外科手术切除。
(3)间质瘤经内镜下微创切除或者外科手术切除后,需根据病理结果,再评估是否需要追加靶向等治疗,或者定期随访。
(4)对于不可切除的恶性胃肠间质瘤,分子靶向药物是首选治疗,在药物治疗过程中进行动态评估,在靶向药物治疗后达到疾病部分缓解或稳定状态,再评估是否可以外科手术切除。
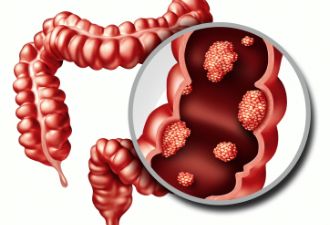
胃肠间质瘤(gastrointestinal stromal tumor,GIST),是胃肠道最常见的间叶源性肿瘤,其生物学行为多样,可以从良性到恶性。
胃肠间质瘤(gastrointestinal stromal tumor,GIST),是胃肠道最常见的间叶源性肿瘤,其生物学行为多样,可以从良性到恶性。
胃肠间质瘤该如何治疗?
(一)对于胃间质瘤来说:
1、胃间质瘤直径大于2cm,需要手术治疗;
2、胃间质瘤直径小于2cm,但合并有临床表现(如肿瘤出血及溃疡形成)或超声胃镜提示:边界不规则、溃疡、囊腔、强回声和回声不均匀等高危因素的病人,建议积极行外科手术治疗;
3、胃间质瘤直径小于2cm,不合并有临床表现(如肿瘤出血及溃疡形成)或超声胃镜提示没有高危因素的病人,可以随访、观察;
随访时间间隔一般是6-12个月,复查超声胃镜或者腹部增强 CT(初次CT检查时可以从片子上看到病灶者,复查时可以做腹部增强CT)。
(二)对于长在十二指肠、空肠、回肠及结直肠等部位的小间质瘤(直径<2cm的间质瘤,称为小间质瘤),一经发现建议尽早完整切除。
(三)对于原发可以切除干净的胃间质瘤,同时不需要联合器官切除且不严重影响器官功能的原发局限性胃肠间质瘤,首选治疗方法是外科手术完整切除。
手术方式可以选择腹腔镜手术,目前推荐腹腔镜用于肿瘤直径≤5 cm 且位于胃前壁、大弯侧的。手术要求完整、不破裂切除病灶。
手术后根据危险度分级等决定是否行辅助治疗

评估具有低危、极低危复发风险的患者手术后不需要辅助治疗
评估具有中、高危复发风险的患者手术后需要辅助治疗
推荐伊马替尼辅助治疗的剂量均为 400 mg/d
①中度复发风险:
▲非胃(主要为小肠、结直肠)来源的GIST,伊马替尼辅助治疗3年
▲胃来源的 GIST,伊马替尼辅助治疗 1 年
②高度复发风险:胃来源或非胃来源的GIST,辅助治疗时间至少 3年
(四)如果手术可能切不干净(R0)的间质瘤,或者需要联合器官切除,或者可完整切除但手术风险较大者,对特殊部位(食管胃结合部、十二指肠及低位直肠等)直径较大的原发间质瘤,直接手术易损害重要器官功能 ,可以术前口服伊马替尼治疗。
术前治疗开始前,须行病理活检明确诊断,并推荐进行基因检测。
伊马替尼初始剂量 400mg/d,一般认为术前治疗6-12个月施行手术比较适宜。
术前 1~2 周停用分子靶向药物。
期间每 3 个月进行影像学检查,具体有病情变化者,还要具体分析。
(五)转移复发/不可切除胃肠间质瘤的治疗
伊马替尼是转移复发 / 不可切除胃肠间质瘤的一线治疗药物,一般主张初始推荐剂量为 400 mg/d。
对于标准剂量的伊马替尼治疗后出现广泛进展者, 建议换用舒尼替尼或选择伊马替尼增加剂量治疗。
胃肠间质瘤该如何随访复查?
(一)随访频率
低危、极低危的病人:通常推荐术后每 6 个月,进行一次随访复查,持续 5年;
中、高危的病人:通常推荐术后每 3个月,进行一次随访复查,持续3年;之后每 6个月 进行一次随访复查,持续 2 年;手术5 年之后每年随访 1 次;
晚期病人:每 3个月,进行一次随访复查。
(二)随访内容:复查项目应包括血常规、生化及腹、盆腔增强CT或MRI。
胃上长的东西,就是胃间质瘤吗?
胃肠间质瘤(gastrointestinal stromal tumor,GIST),是胃肠道最常见的间叶源性肿瘤,其生物学行为多样,可以从良性到恶性。
蛋白层面进行检测:免疫组化检测通常表达 CD117 和 DOG1 阳性;
基因层面进行检测:大多数病例具有 c-kit 或血小板源性生长因子受体 α 多肽(platelet derived growth factor receptor alpha,PDGFRA)基因活化突变。基因检测应至少包括 ckit 基因第 9、11、13、17 号外显子及 PDGFRA 基因第12、18号外显子。
胃间质瘤都有哪些临床表现、诊断?
临床表现多样且缺乏特异性。主要取决于肿瘤大小、部位及生长方式,常见的临床表现包括腹部不适、腹痛、黑便及腹部包块等,也有一部分病人因体检或诊治其他疾病偶然发现,胃镜或者CT提示黏膜下隆起性病变。
胃和小肠是最常见的原发部位,结直肠、食管及胃肠道外GIST比较少见。
常用检查方式包括内镜、超声内镜、CT 及MRI 等。
不常规推荐进行活组织检查,可直接手术切除;只有术前检查考虑复发转移、原发不可切除或特殊部位需术前治疗的胃肠间质瘤应进行组织活检,目的是明确肿瘤性质及基因分型,进而指导靶向药物治疗。
随着目前研究越来越多,药物发展迅速,建议间质瘤患者行基因检测,更好地指导临床的用药。
了解更多肿瘤相关健康知识→敬请关注周医生

《我不是药神》的故事是闪现回到 2002 年。一个普通得不能再普通的小人物程勇(徐峥饰),经营着一家没什么生意起色的印度神油保健品店。和大多数小人物一样,却遭遇了他迈不过去的大事件:父亲病危、穷得叮当响,同时前妻还要带走儿子。混的是值得大多数人同情的那种惨。

不过偶然的一下子,他发现了国内的某些白血病患者,需要依靠一种叫作「格列宁」的天价药物续命,而且仅此一种药才能续命,「格列宁」,简直是「孤儿药」中的战斗机。而这种药,却在神奇的国度印度,可以以几十分之一的极低的价格买入印度药厂生产的仿制药。尽管和进口的「格列宁」这种原研药比起来,印度产的「格列宁」的安全性和疗效都没得到国内专家的认同,甚至连购入途径都是「徘徊在合法边缘」。但是需求就是市场,这个小人物程勇就做起了这个「代购」生意。
所以在药品买卖「代购」的长篇故事里,穿插着白血病人绝望的抓稻草的身影,穿插着侦办假药警察的为难,本来只是只关金钱的故事,却因为掺入了生死、法与情的成分,变成了一个复杂的故事。 一部带着泪的「喜剧」,一部令人期待会在中国影史画上浓墨重彩一笔的电影。
而更为魔幻的事,这个故事真正的发生在我们同生的这片土地。而「格列宁」,正是明星药「格列卫」的化名。这个魔幻的故事,的的确确的悄无声息地发生在格列卫这款明星药身上。 格列卫,化学名 imatinib(inn,伊马替尼),是诺华制药公司(novartis)研发的一种针对酪氨酸激酶 bcr-abl 的分子靶向药物。在欧、澳及南美洲被称为「glivec」,在美国则被称为「gleevec」。如果我没记错,也是全球第一款上市的分子靶向药。曾经对于很多人甚至医护人员而言,也是不甚熟悉。 这款药,不仅用于治疗费城染色体阳性的慢性髓性白血病(ph+cml)的慢性期、加速期或急变期,还可以以及用于治疗不能切除或发生转移的恶性胃肠道间质肿瘤(gist)的成人患者。 所以每次收治的胃肠道间质瘤患者后,同他们讲起格列卫,常常会这样开头:「还记得原来那个新闻讲有个人跑到印度给患者买印度仿制药的故事么,他去买的药,就是这个格列卫」从现在开始,恐怕我再也不需要这么费劲的去费劲的解释这个明星药了。
虽然没有胃癌那么「名声在外」,可胃肠间质瘤,既然也是恶性肿瘤的一种,也具有其独特的「令人闻风丧胆」的坏能力。胃肠间质瘤恶性程度不一,但都有可能发生复发或转移,以血运转移和胸腔广泛转移最常见,较少转移到淋巴结、骨、肺。

更让人心累的是,间质瘤在治疗方面,胃肠间质瘤对传统的放疗、化疗不敏感,而且容易复发。大多数那些普通的抗肿瘤药,对他都是束手无策。但值得庆幸的是,自从这个格列卫(伊马替尼)问世以后,身患胃肠道间质瘤的患者也有了控制病情的机会。 对于大多数病人,先依靠外科手术进行间质瘤的根治切除。但是,经彻底手术切除,胃肠间质瘤复发率仍高达 80%。这与它细胞生长活跃及患者本身的基因突变有关。即使是晚期转移复发的胃肠间质瘤患者,也可采取伊马替尼靶向治疗,把肿瘤变成一个慢性状态,控制肿瘤进展,延长患者生存期。 因此,尽管胃肠间质瘤让不少患者在一开始万念俱灰,但如果进行手术切除,术后采用靶向药物治疗,定期复查,绝大多数患者可以回归工作岗位,像正常人一样地生活。
不过令人振奋的是,大家看着《我不是药神》的电影,对高昂的天价格列卫咂舌感叹,但殊不知,格列卫在最近几年,在国内的许多省份,以及陆续进入到医保里面。而且国内同样也有一些药企获得批准,可以生产上市格列卫的国产仿制药。
所以近年来,我们诊治的间质瘤患者,基本有药可用。以前动辄一盒两万多的吃不了几天的花费,现在基本一年的费用在医保的帮助下可以控制在万把多块钱。 不过依然还是很想给这部电影点赞,记录了这样一群热爱生命不言放弃的人抗争的身影,给大家不只是感官上的震撼,以及更多深入复杂人性的思考。 也许从此之后,格列卫不再遥不可及,可历史值得铭记。
当地时间9月12日,FDA批准首个皮下注射抗PD1免疫疗法,用于特定类型肺癌、肝癌、黑色素瘤、软组织癌。这个皮下疗法组合包括两部分组成:一部分是阿替利珠单抗;另一个是 Halozyme公司自己研制的重组人透明质酸酶rHuPH20[1]。
皮下疗法应用Halozyme的ENHANZE药物递送技术,可将原本30mim的静注时间缩短为7min。让我们一起来看支撑这项研究背后的临床试验结果。
相似的血药浓度、有效性和安全性
该获批基于一项随机多中心 Ib/III 期的试验,研究的主要目的要看看阿替利珠单抗这个药,打在皮下和打在静脉里在一些特定患者身上效果有什么不一样。这些特定患者为局部晚期或者已经转移的非小细胞肺癌患者,他们之前没做过癌症免疫治疗,而且之前用含铂类的药治疗也没效果。
试验的主要结果显示,皮下注射atezolizumab和静脉注射 atezolizumab 的几何平均比值 (GMR) 为1.05 (90% CI,0.88-1.24),AUC 0-21 天为 0.87 (90% CI,0.83-0.92),这表明皮下注射可以达静脉注射相当的血药浓度。有效性方面,皮下和静脉的总缓解率 (ORR) 、无进展生存期 (PFS) 和总生存期 (OS) 数据相似[2]。
安全性方面,最常见的任何级别不良反应是疲劳、肌肉骨骼疼痛、咳嗽、呼吸困难和食欲下降。
关于阿替利珠单抗
根据FDA获批信息,该药适应证如下,本次的获批主要是制剂类型的不同。
适应证[3]
1. 非小细胞肺癌(NSCLC):
- 单药用于经手术及含铂化疗后的Ⅱ至ⅢA期NSCLC成人患者辅助治疗(肿瘤细胞PD-L1表达≥1%)。
- 单药一线治疗高表达PD-L1、无EGFR或ALK畸变的转移性NSCLC成人患者。
- 与贝伐珠单抗等联合用于无EGFR或ALK畸变的转移性非鳞状NSCLC成人患者一线治疗。
- 单药治疗含铂化疗期间或后进展的转移性NSCLC成人患者(有EGFR或ALK畸变患者应在对应FDA批准疗法进展后使用)。
2. 小细胞肺癌(SCLC):与卡铂和依托泊苷联合用于广泛期SCLC成人患者一线治疗。
3. 肝细胞癌(HCC):与贝伐珠单抗联合用于未接受全身治疗的不可切除或转移性HCC成人患者。
4. 黑色素瘤:与考比替尼和维莫非尼联合用于BRAF V600突变阳性的不可切除或转移性黑色素瘤成人患者。
5. 肺泡软组织肉瘤(ASPS):单药用于2岁及以上不可切除或转移性ASPS成人及儿童患者。
关于Halozyme[4]
Halozyme 是一家生物制药公司,致力于提供颠覆性解决方案,以改善新兴和成熟疗法的患者体验和结果。作为采用专有酶 rHuPH20 的 ENHANZE药物输送技术的创新者,Halozyme 经过商业验证的解决方案用于促进注射药物和液体的皮下输送,目的是通过快速皮下输送和减轻治疗负担来改善患者体验。
Halozyme 已在全球 100 多个市场的 8 种商业化产品中触及超过 800,000名患者的上市后使用,其 ENHANZE技术已授权给领先的制药和生物技术公司,包括罗氏、武田、辉瑞、杨森、艾伯维、礼来等。
参考文献:
1.Halozyme Announces FDA Approval of Roche's Tecentriq Hybreza™ With ENHANZE® for Multiple Types of Cancer
2.FDA Approves Subcutaneous Atezolizumab and Hyaluronidase-tqjs for Use in All Indications of IV Atezolizumab
3.Product information:.TECENTRIQ- atezolizumab injection, solution
4.https://ir.halozyme.com/news/news-details/2024/Halozyme-Announces-FDA-Approval-of-Roches-Tecentriq-Hybreza-With-ENHANZE-for-Multiple-Types-of-Cancer/default.aspx

2024年9月10日,诺和诺德一项减肥研究登顶《新英格兰医学杂志(NEJM)》,公布了胰高血糖素样肽-1(GLP-1)受体激动剂利拉鲁肽在儿童中的减重数据:利拉鲁肽组近一半参与者BMI降低 5%以上。让我们一起来看看这项儿童减肥药的研究。
火锅烤肉、奶茶饮料等诱惑层出不穷,让不做选择的成年人慢慢走上了肥胖之路。而我们祖国的花朵,面对这些,也茁壮成长成了“小胖子”。据统计,我国6~17岁、6岁以下儿童和青少年超重/肥胖率分别达到19.0%和10.4%[1]。
孩子肥胖与很多疾病息息相关,如2型糖尿病、脂肪性肝炎、多种癌症。除了引导健康饮食和规律的生活方式[2],一些药物选择对于超重儿童提供了选择。
虽然经常提到司美格鲁肽、利拉鲁肽,但是其在儿童中的安全性一直引人担忧,于是就有了NEJM这个研究。这个研究是一项双盲随机对照试验。试验分为利拉鲁肽组和对照组,参与者每日一次注射药物,在4-5周内每周剂量递增,直至达到最终剂量[3]。试验有效性结果主要看参与者BMI变化百分比。
共有 82 名参与者接受了随机分组;56 人被分配至利拉鲁肽组,26 人被分配至安慰剂组。
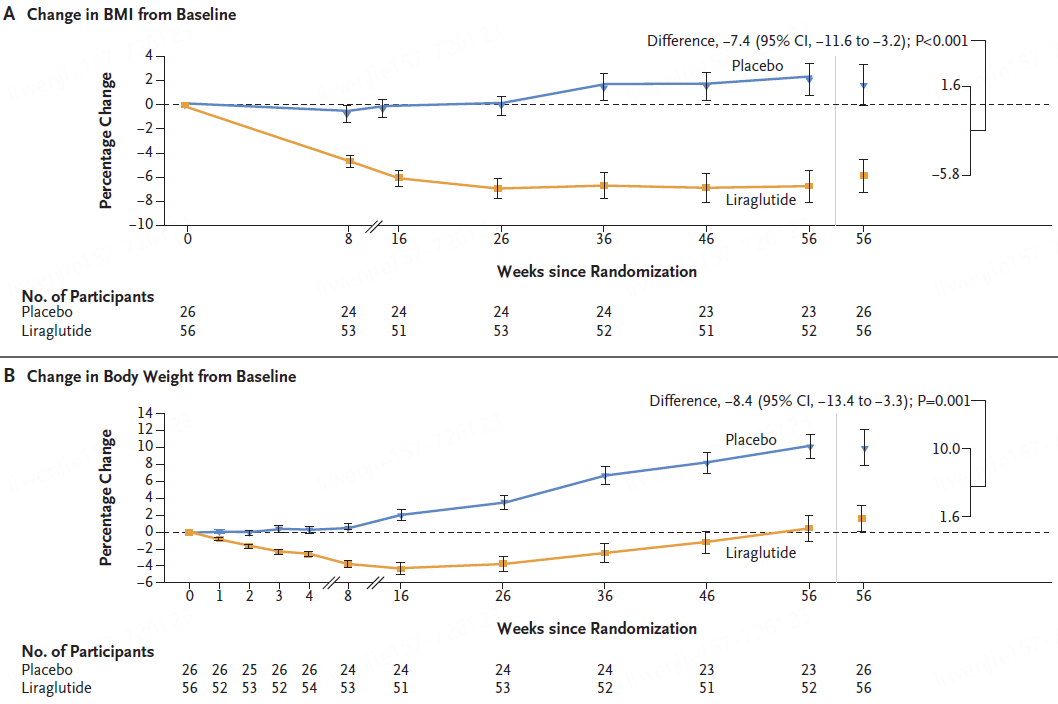
试验结果表明,在第 56 周时,利拉鲁肽组BMI较基线的平均变化百分比为 - 5.8%,安慰剂组为 1.6%。
利拉鲁肽组体重的平均变化百分比为 1.6%,安慰剂组为 10.0%。
利拉鲁肽组46%的参与者BMI降低至少5%,而安慰剂组为 9%(调整后的优势比为 6.3 [95% CI,1.4 至 28.8];P = 0.02)。
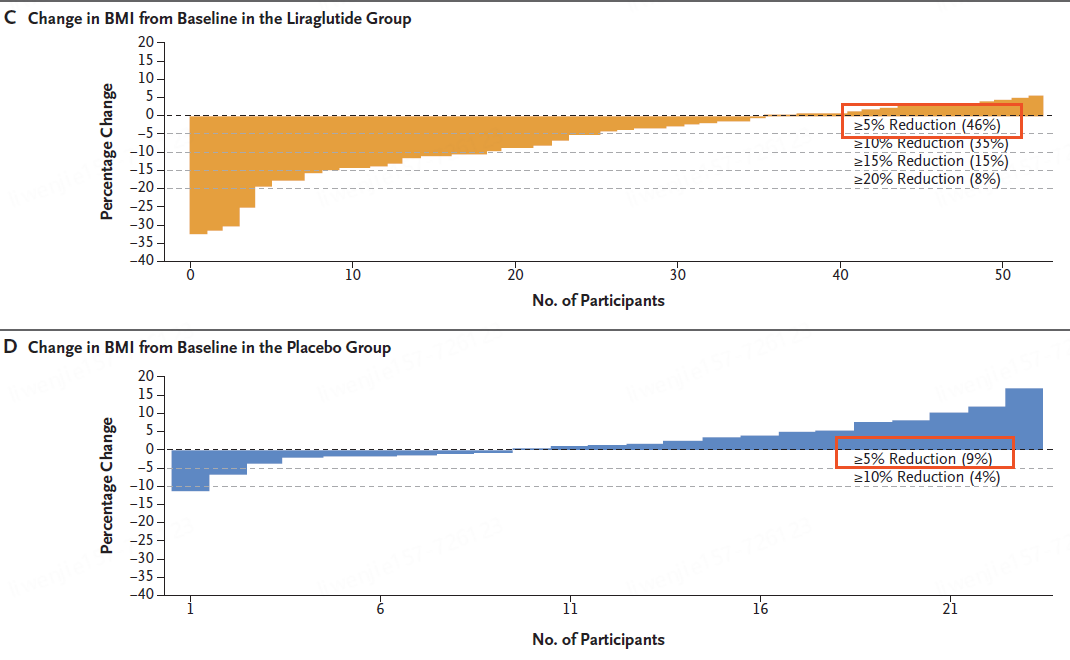
利拉鲁肽组和安慰剂组分别有 89% 和 88% 的参与者发生不良事件。胃肠道不良事件在利拉鲁肽组更为常见(80% vs 54%);利拉鲁肽组和安慰剂组分别有 12% 和 8% 的参与者报告了严重不良事件。
关于利拉鲁肽
利拉鲁肽(VICTOZA)的已获批适应症包括:
作为饮食和运动的辅助手段,用于改善 10 岁及以上成人和儿童 2 型糖尿病患者的血糖控制。
降低已确诊心血管疾病的成人 2 型糖尿病患者发生主要不良心血管事件(心血管死亡、非致命性心肌梗死或非致命性中风)的风险。
说明书也也发布了黑框警告信息:
本品禁用于有甲状腺髓样癌个人或家族史的患者以及患有 2 型多发性内分泌腺瘤综合征(MEN 2)的患者。应告知患者使用本品可能存在的甲状腺髓样癌风险,甲状腺肿瘤的症状包括颈部肿块、吞咽困难、呼吸困难、持续性声音嘶哑。
关于诺和诺德[5]
诺和诺德是一家全球领先的医疗保健公司,成立于1923年,总部位于丹麦。其的宗旨是在糖尿病领域的传统基础上,推动变革以战胜严重的慢性疾病。诺和诺德通过开创科学突破、扩大药品的可及性以及致力于预防并最终治愈疾病来实现这一目标。诺和诺德在80个国家拥有约 69,000名员工,产品在大约170个国家销售。
参考文献:
1.中国超重/肥胖医学营养治疗指南(2021)
2.Fox CK, Barrientos-Pérez M, Bomberg EM, Dcruz J, Gies I, Harder-Lauridsen NM, Jalaludin MY, Sahu K, Weimers P, Zueger T, Arslanian S; SCALE Kids Trial Group. Liraglutide for Children 6 to <12 Years of Age with Obesity - A Randomized Trial. N Engl J Med. 2024 Sep 10. doi: 10.1056/NEJMoa2407379. Epub ahead of print. PMID: 39258838.
3.SCALE KIDS: Research Study to Look at How Well a New Medicine is at Lowering Weight in Children With Obesity
4.Product information:VICTOZA- liraglutide injection
5.https://www.novonordisk.com/

在 2024 年世界肺癌大会上,一项关于局部晚期或转移性 ROS1 阳性非小细胞肺癌(NSCLC)的试验公布了数据,这是一项多中心Ⅱ期试验,试验药物是新一代酪氨酸激酶抑制剂(ROS1 TKI)taletrectinib,在试验中的代码是AB - 106[1]。
总体缓解率达85.2%,亚洲患者达87.9%
试验的主要结果是客观缓解率(ORR),最长时间为 4 年,入组患者根据既往接收ROS1 TKI治疗的情况分为两组,一组既往胃接受,另外一组接受过[2]。从试验的结果来看,未接收过这类药物治疗ORR比例更好,达85.2%。详细数据如下所示:
- 先前未接受过 ROS1 TKI 治疗的患者(n = 54),经过中位随访 15.8 个月(范围 3.6 - 29.8),taletrectinib 展现出了较好疗效,确认总体缓解率(cORR)高达 85.2%(95%CI,72.88% - 93.38%)。值得一提的是,亚洲患者(n = 33)与非亚洲地区患者的 cORR 相近,分别为 87.9%(95%,71.80% - 96.60%)和 81.0%(95%CI,58.09% - 94.55%)。
- 先前接受过 ROS1 TKI 治疗的患者中(n = 47),中位随访 15.7 个月(范围 3.9 - 29.8),taletrectinib 的 cORR 也达到了 61.7%(95%CI,46.38% - 75.49%)。亚洲患者(n = 21)与其他地区患者(n = 26)的 cORR 分别为 57.1%(95%CI,34.02% - 78.18%)和 65.4%(95%,44.33% - 62.79%)。
- 在基线时有可测量脑转移的患者中,评估颅内反应显示,初治组(n = 9)的颅内客观缓解率(IC - ORR)为 66.7%(95%CI,29.93% - 92.51%),其中完全缓解(CR)率为 22.2%,部分缓解(PR)率为 44.4%[1]。
最常见不良反应为肝药酶升高
导致剂量减少的治疗出现的不良事件(TEAEs)发生在 37.1%的患者中,其中 16.4%是由于肝酶升高。导致治疗中断的 TEAEs 报告在 7.5%的患者中;其中 1.3%被认为与治疗相关。没有报告导致死亡的 TEAEs。
其他不良反应发生率如下:
- 至少 15%的患者(n = 159)中最常见的 TEAEs 是丙氨酸氨基转移酶升高(任何级别,67.9%;≥3 级,15.1%),天冬氨酸氨基转移酶升高(67.3%;6.9%),腹泻(56.6%;0.6%),恶心(51.6%;1.9%),呕吐(33.3%;1.3%),便秘(25.2%;0.0%),贫血(20.1%;4.4%),味觉障碍(19.5%;0.0%),血肌酸磷酸激酶升高(18.2%;3.8%),头晕(17.0%;0.0%)和 QT 延长(15.1%;3.1%)[2]。
Taletrectinib是何许药物?
最后再回到taletrectinib,这个药作为新一代 ROS1 TKI 药物,在TRUST - II 试验中表现确实亮眼。
由于对 ROS1+肿瘤有着更高的效力,taltrectinib 能够提升患者无进展生存期(PFS),同时这个药物的中枢神经系统(CNS)渗透率也更高,药理机制上,可以有选择性地抑制 ROS1 野生型以及其对 TRKB 的耐药突变;与其他 ROS1+抑制剂相比,taltrectinib 的安全性更优,并且与中枢神经系统相关的不良事件(AE)极少[3]。
参考文献
1.Taletrectinib Shows Consistent Efficacy, Safety in ROS1+ NSCLC Independent of Prior TKI Exposure
2.Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)
3.Nagasaka M, Brazel D, Ou SI. Taletrectinib for the treatment ofROS-1positive non-small cell lung cancer: a drug evaluation of phase I and II data. Expert Opin Investig Drugs. 2024 Feb;33(2):79-84. doi: 10.1080/13543784.2024.2305131. Epub 2024 Jan 29. PMID: 38224083.

近期Merck宣布其正在进行的IDeate-Lung01 Ⅱ 期试验结果积极,试验主角ifinatamab deruxtecan(I-DXd)用于治疗广泛期小细胞肺癌(ES-SCLC),在入组患者中,12mg/kg I-DXd 1的客观缓解率(ORR)为 54.8%[1]。

IDeate-Lung01是一个多中心随机开放研究,主要结果:在广泛期小细胞肺癌(ES-SCLC)患者中,使用I-DXd治疗后的客观缓解率(ORR)百分比,试验最长约 36 个月[2]。
这里的客观缓解率就是在参与试验的人中,达到最佳总体缓解的人所占的百分比,“总体缓解”指的是经确认的完全缓解或者部分缓解。对于所有的靶病灶、非靶病灶和新出现的病灶来说,完全缓解就是所有的病灶都消失了;部分缓解就是所有病灶直径的总和至少减少了 30%。
疾病控制率高达90.5%,客观缓释率达54.8%
经盲态独立中心审查(BICR)评估,在接受I-DXd治疗的广泛期小细胞肺癌患者中,12mg/kg 队列(n = 42)的确认客观缓解率(ORR)为 54.8%(95%置信区间:38.7 - 70.2),8mg/kg 队列(n = 46)为 26.1%(95%置信区间:14.3 - 41.1)[1]。

在 12mg/kg 队列中观察到 23 个部分缓解(PR)。在 8mg/kg 队列中观察到 1 个完全缓解(CR)和 11 个部分缓解。
在 12mg/kg 和 8mg/kg 队列中,分别观察到中位缓解持续时间(DoR)为 4.2 个月(95%置信区间:3.5 - 7.0)和 7.9 个月(95%置信区间:4.1 - 未确定),疾病控制率(DCR)为 90.5%(95%置信区间:77.4 - 97.3)和 80.4%(95%置信区间:66.1 - 90.6)。
12mg/kg 剂量的中位治疗持续时间为 4.7 个月(0.03 - 15.2),8mg/kg 剂量为 3.5 个月(0.03 - 13.9)。在 12mg/kg 和 8mg/kg 队列中,分别观察到中位无进展生存期(PFS)为 5.5 个月(95%置信区间:4.2 - 6.7)和 4.2 个月(95%置信区间:2.8 - 5.6),中位总生存期(OS)为 11.8 个月(95%置信区间:8.9 - 15.3)和 9.4 个月(95%置信区间:7.8 - 15.9)。
12mg/kg 剂量已被选定用于试验的剂量第二部分。截至 2024 年 4 月数据截止时,12mg/kg 队列的中位随访时间为 15.3 个月(95%置信区间:13.6 - 16.2),8mg/kg 队列为 14.6 个月(95%置信区间:13.4 - 16.5)。 在有脑靶病灶的患者中,经中枢神经系统(CNS)BICR 评估,在 12mg/kg 队列(n = 10)和 8mg/kg 队列(n = 6)中分别观察到颅内客观缓解率为 50.0%(95%置信区间:18.7 - 81.3)和 66.7%(95%置信区间:22.3 - 95.7)。在这些患者中,每个队列中均观察到两个颅内完全缓解。
安全性数据
12mg/kg 队列中 50.0% 患者发生 3 级或更高等级治疗中出现的不良事件,8mg/kg 队列中 对应比例达43.5% 。两种剂量下常见治疗相关不良事件有恶心、食欲下降、贫血、中性粒细胞减少、白细胞减少和乏力等。
12mg/kg 和 8mg/kg 剂量下分别有五例和四例间质性肺病、肺炎事件被确认为与治疗相关,多数为低级别。12mg/kg 和 8mg/kg 队列中因不良事件导致的治疗中断分别为 16.7% 和 6.5%。
关于I-Dxd
I-Dxd是一种首创 B7-H3 靶向抗体药物偶联物(ADC),与第一三共开发的其他抗体药物偶联物(一样,向癌细胞递送相同的毒性有效载荷。I-Dxd靶向一种B7-H3 的蛋白质,该蛋白质在包括前列腺癌和食管癌在内的多种癌症中过度表达[3]。
参考来源:
1.Ifinatamab Deruxtecan Continues to Demonstrate Promising Objective Response Rates in Patients with Extensive-Stage Small Cell Lung Cancer in IDeate-Lung01 Phase 2 Trial
2.Ifinatamab Deruxtecan (I-DXd) in Subjects With Pretreated Extensive-Stage Small Cell Lung Cancer (ES-SCLC) (IDeate-Lung01)
3.Merck, Daiichi Sankyo detail Phase 2 ADC results in small cell lung cancer, bispecific combo strategy

2024年9月5日,Vor Bio公布了其1/2 期 VBP101 试验的临床数据,VBP101主要评估干细胞疗法 tremtelectogene empogeditemcel(trem - cel)与 CD33 靶向抗体偶联药物(ADC)Mylotarg 联用用于治疗复发/难治性急性髓系白血病(AML)患者的效果[1]。
干细胞移植后白血病复发风险
虽然患者进行了造血干细胞移植(HCT),但高风险急性髓系白血病(AML)和骨髓增生异常综合征(MDS)仍经常复发。HCT后的靶向治疗可减少复发,但会对植入细胞产生毒性限制。
VOR33 是一种异基因 CRISPR/Cas9 基因组编辑的造血干细胞和祖细胞(HSPC)治疗产品,缺乏 CD33 蛋白,这个试验正是对 HCT 后复发风险高的 CD33 + AML 或 MDS 患者进行研究,以便在 HCT 后使用 Mylotar靶向残留的 CD33 + 急性 AML 细胞,从而避免对植入的 VOR33 细胞产生毒性。
参与者将接受HCT植入,植入后将接受 Mylotarg治疗,治疗周期≤4个周期。评估 VOR33 安全性的主要终点是 28 天成功植入的发生率。研究将首先评估递增 Mylotarg剂量水平的安全性,以确定最大耐受剂量(MTD)和推荐的 2 期剂量(RP2D)。后续将进一步扩大参与者数量,以评估 Mylotarg的 RP2D[2]。
100%植入,无复发生存期改善
公布的数据包括 18 名接受 trem - cel 治疗的患者,截至 2024 年 7 月 19 日的数据截止日期,其中 10 名患者接受了 Mylotarg 治疗。详细数据如下[1]:
可靠的植入效果:100% 的患者实现了初级中性粒细胞植入(中位时间为 9 天)和强劲的血小板恢复(中位时间为 16.5 天)。在第 28 天,CD33 编辑效率高(中位数为 89%,范围为 71 - 94%),并且实现了完全的髓系嵌合。
血液系统的屏障:不同 Mylotarg 剂量( 0.5、1和 2mg/平方米)下,中性粒细胞和血小板计数保持稳定。
Mylotarg 治疗指数的拓宽:药物暴露由 AUC 表示,与疗效相关,与标记的 Mylotarg 剂量一致;最大浓度由 Cmax 远低于已知的毒性范围。
早期证据表明患者受益:与已发表的高危 AML 对照者相比,在无复发生存期方面,患者受益。
VBP301 研究将继续招募患者,继续关注其试验目的。预计今年年底,Vor Bio计划与FDA讨论trem - cel与Mylotarg联合用药的关键试验设计。
Vor Bio 是一家扔处于临床阶段的细胞和基因组工程公司,其多条管线扔处于临床研究阶段(详见图片),旨在通过对造血干细胞进行工程改造,使移植后的靶向治疗成为可能,从而改变血癌患者的护理标准。
参考文献:
1.New Clinical Data Validates Vor Bio’s Approach of Using Shielded Transplants to Deliver Targeted Therapies
2.Allogeneic Engineered Hematopoietic Stem Cell Transplant (HCT) Lacking the CD33 Protein, and Post-HCT Treatment With Mylotarg, for Patients With CD33+ AML or MDS
四川科伦博泰生物医药股份有限公司(以下简称“科伦博泰”)在2024ESMO年会上公布三项临床研究结果。
1、芦康沙妥珠单抗(sac-TMT,前称SKB264/MK-2870)联合帕博利珠单抗用于复发或转移性宫颈癌(CC)患者的疗效及安全性。
2、芦康沙妥珠单抗(sac-TMT)单药疗法用于既往接受过治疗的晚期子宫内膜癌(EC)及卵巢癌(OC)患者的2期研究的安全性及疗效。
3、在芦康沙妥珠单抗(sac-TMT)对比化疗用于既往接受过治疗的晚期三阴性乳腺癌(TNBC)患者的3期研究(OptiTROP-Breast01)中,对既往接受过或未接受过 PD-(L)1 抑制剂治疗的患者的探索性分析。
CC
在含铂双药化疗中或之后出现疾病进展且接受过不超过2种针对复发或转移性(R/M)疾病系统治疗(允许使用PD-(L)1抑制剂治疗)的R/M CC患者入组并于安全导入期接受每2周一次3或5 mg/kg剂量的芦康沙妥珠单抗(sac-TMT)联合每6周一次400 mg剂量的帕博利珠单抗治疗评估,并在剂量拓展阶段探索到视为良好耐受的剂量。
截至2024年3月25日的数据截止日期,38名患者接受了治疗并接受为期至少17周的随访或两次肿瘤评估(3名患者接受3mg/kg剂量的芦康沙妥珠单抗(sac-TMT)治疗,35名患者接受5mg/kg剂量的芦康沙妥珠单抗(sac-TMT)治疗)。中位随访时间为6.2个月。患者的中位年龄为52岁。76.3%的患者有鳞状组织,47.4%既往接受过二线治疗,52.6%既往接受过贝伐珠单抗治疗,42.1%既往接受过抗PD-1疗法。ORR为57.9%(22/38,19例(50%)已确认),其中3例完全缓解。中位持续缓解时间(DoR)尚未达到,6个月DoR率为82.1%。在接受抗PD-1疗法的患者中亦观察到缓解(ORR为68.8%,11/16)。中位PFS尚未达到,6个月PFS率为65.7%。
47.4%的患者发生≥3级TRAE。最常见的≥3级TRAE为中性粒细胞计数减少(23.7%)、贫血(21.1%)及白细胞计数减少(15.8%)。44.7%的患者出现导致芦康沙妥珠单抗(sac-TMT)剂量降低的TRAE,1例患者(2.6%)出现芦康沙妥珠单抗(sac-TMT)停药的TRAE。并无导致两种药物停药的TRAE。
默沙东发起的一项评估芦康沙妥珠单抗(sac-TMT)单药疗法对比研究者选择方案(TPC)用于二线治疗复发或转移性CC患者的3期全球研究(NCT06459180)正在进行中。
EC及OC
既往接受过含铂化疗的分别两个队列的晚期EC及OC患者每2周接受一次5 mg/kg剂量的芦康沙妥珠单抗(sac-TMT),直至发生疾病进展、不可耐受的毒性或撤回同意。TROP2表达采用半定量H评分方法进行评分,并将分界点设置为200。
截至2024年3月5日的数据截止日期,共有44名患者入组EC队列,中位随访时间为7.2个月。52.3%的患者既往接受过二线或以上的治疗。客观缓解率(ORR)为34.1%(15/44,12例已确认),疾病控制率(DCR)为75%。中位无进展生存期(PFS)为5.7个月(95%置信区间(CI):3.7,9.4),6个月PFS率为47.5%。对于TROP2免疫组织化学(IHC) H评分>200的患者(n=12),ORR为41.7%(5/12,3例已确认),而对于H评分≤200的患者(n=28),ORR为35.7%(10/28,9例已确认)。共有40名患者入组OC队列,中位随访时间为28.2个月。所有患者既往均接受过二线或以上的治疗(80%的患者既往接受过三线或以上的治疗)且87.5%的患者对含铂药物耐药。ORR为40%(16/40,14例已确认),DCR为75%。中位PFS为6.0个月(95% CI:3.9,7.3),中位总生存期(OS)为16.5个月(95% CI:10.7,无法估计(NE))。在TROP2 IHC H评分>200的患者(n=13)中,ORR为61.5%(8/13,7例已确认),在H评分≤200的患者(n=22)中,ORR为27.3%(6/22,6例已确认)。在对含铂药物耐药的患者中(n=35),中位PFS为6.0个月(95% CI:5.3,7.3),中位OS为16.1个月(95% CI:10.5,NE)。
分别有72.7%及67.5%的EC和OC患者发生≥3级TRAE。最常见的≥3级TRAE(≥15%)(EC及OC)为中性粒细胞计数减少(43.2%及30.0%)、白细胞计数减少(40.9%及22.5%)、贫血(29.5%及35.0%)及口腔炎(29.5%及35.0%)。分别有1例(2.3%)和5例(12.5%)的EC和OC患者出现导致停药的TRAE。
一项由默沙东发起的评估芦康沙妥珠单抗(sac-TMT)单药疗法用于治疗既往接受过含铂化疗和免疫治疗的EC患者的3期全球研究(NCT06132958)正在进行中。
TNBC
既往接受过≥2种治疗(其中至少1种治疗针对转移性阶段)的局部复发或转移性TNBC患者随机接受芦康沙妥珠单抗(sac-TMT)或研究者选择方案。主要终点为盲态独立中心审阅委员会(BICR)评估的PFS。
截至2023年11月30日,接受芦康沙妥珠单抗(sac-TMT)治疗的患者中有24.6%(32/130)既往接受过PD-(L)1抑制剂治疗,接受TPC治疗的患者中有27.1%(36/133)既往接受过PD-(L)1抑制剂治疗。在该亚组中观察到芦康沙妥珠单抗(sac-TMT)对比TPC具有临床获益。BICR评估的中位PFS为5.6个月对比2.7个月(HR 0.31;95% CI 0.17-0.54),BICR评估的接受芦康沙妥珠单抗(sac-TMT)治疗患者的ORR为56.3%,而接受TPC治疗患者的ORR为5.6%。对于既往未接受过PD-(L)1抑制剂治疗的患者,芦康沙妥珠单抗(sac-TMT)对比TPC在疗效方面有类似的改善。中位PFS为7.2个月对比2.3个月(HR 0.34;95% CI 0.23-0.48),ORR为41.8%对比14.4%。
芦康沙妥珠单抗(sac-TMT)组中既往接受过或未接受过PD-(L)1抑制剂治疗的患者之间的安全性数据相似。
芦康沙妥珠单抗(sac-TMT)用于既往接受过治疗的局部复发或转移性TNBC患者的3期研究(OptiTROP-Breast01)结果已于2024年6月2日在2024年ASCO年会上进行了公布,详情载于本公司2024年中期业绩公告及日期为2024年5月24日的公告。
一项由默沙东发起的评估芦康沙妥珠单抗(sac-TMT)联合帕博利珠单抗对比TPC用于既往接受过新辅助治疗且未达到病理学完全缓解(pCR)的TNBC患者的3期全球研究(NCT06393374),及本公司发起的一项评估芦康沙妥珠单抗(sac-TMT)在中国用于一线治疗不可手术切除的局部晚期、复发或转移性PD-L1阴性TNBC患者的3期研究(NCT06279364)均正在进行中。
于2022年5月,本公司授予默沙东(美国新泽西州罗威市默克公司的商号)在大中华区(包括中国内地、香港、澳门以及台湾)以外的所有地区开发、使用、制造及商业化芦康沙妥珠单抗(sac-TMT)的独家权利。可瑞达®为美国新泽西州罗威市默克公司的附属公司Merck Sharp & Dohme LLC的注册商标。
此次芦康沙妥珠单抗(sac-TMT)在ESMO大会公布的针对不同适应症的研究成果,有望进一步满足包括三阴性乳腺癌在内的妇科肿瘤的治疗需求,使更多的临床患者获益。
2024年9月5日,礼来公司官宣QWINT-1和QWINT-3两项关于糖尿病的Ⅲ期试验取得积极进展[1]。
这两项临床试验分别在两类患者中评估胰岛素周制剂efsitora的效果,这两类患者分别是首次使用基础胰岛素(未使用过胰岛素)的 2 型糖尿病患者、从每日注射基础胰岛素换成新药的患者。
从提高患者依从性而言,这项研究有重要意义,既往糖尿病患者每次用餐前都需要提取注射胰岛素,若出去吃饭可能会带来一些不便之处,如果能改为周注射还能控制好血糖,或许是糖尿病患者的福音。一起来看这两项研究积极的进展。
QWINT-1中52周糖化血红蛋白降低1.31%
QWINT-1研究是一项随机对照Ⅲ期试验,研究目的是评估与甘精胰岛素相比,每周使用固定剂量递增的胰岛素 efsitora (LY3209590)在首次使用基础胰岛素治疗的 2 型糖尿病(T2D)成人患者中的疗效和安全性[2]。
研究共纳入796名参与者,研究的主要评估结果是糖化血红蛋白(HbA1c)相对于基线的变化。根据礼来公布的结果,在第 52 周时,efsitora 组HbA1c 降低了 1.31%,HbA1c水平为6.92%;甘精胰岛素组降低了 1.27%,HbA1c水平为6.96%。这个试验主要对比efsitora和甘精胰岛素甘精的非劣效性,即控制血糖效果类似[1]。
QWINT-3中26周糖化血红蛋白降低0.86%
研究共纳入986名参与者,QWINT-3研究是一项多中心随机平行Ⅲ期临床试验,旨在评估 LY3209590 与德谷胰岛素在目前接受基础胰岛素治疗的 2 型糖尿病患者中的疗效和安全性,该研究最多持续86 周[3]。
主要研究结果QWINT-1一样,在第26周时,efsitora 组 HbA1c 降低了 0.86%,而德谷胰岛素组HbA1c 降低了 0.75%,26周时HbA1c水平分别为为 6.93% 和 7.03%。也验证了efsitora 与德谷胰岛素相比的非劣效性[1]。
就二个试验安全性而言,在QWINT - 1中,从试验开始到第52周,低血糖(血糖<54mg/dL)的综合发生率为:efsitora组为0.50,甘精胰岛素组为0.88,efsitora组比甘精胰岛素组低约40%。
在QWINT-3中,从试验开始到第78周,低血糖(血糖<54mg/dL)的综合发生率:efsitora组为0.84,胰岛素德谷组为0.74。
关于LY3209590
胰岛素 Efsitora Alfa(efsitora)基础胰岛素周制剂,是一种融合蛋白,将一种新型的单链胰岛素变体与人 IgG2 Fc 结构域结合在一起。其专门设计用于每周一次的皮下注射,,有可能在整个星期内提供更稳定的血糖水平(更少的血糖变异性)。目前该制剂正处于针对 1 型和 2 型糖尿病成人的 Ⅲ 期开发阶段[1]。
2型糖尿病
糖尿病是一组由多病因引起以高血糖为特征的代谢性疾病,是由于胰岛素分泌和/或利用缺陷引起。长期碳水化合物以及脂肪、蛋白质代谢紊乱可引起多系统损伤,导致眼、肾、神经、心脏、血管等组织器官发生进行性病变、功能减退及衰竭[4]。
我国≥65 岁的老年糖尿病患者数量约为3550万[5],居世界首位,且呈现上升趋势。老年糖尿病患者常伴有多种慢性疾病,多重用药较为普遍,可能影响降糖疗效及增加低血糖风险,患者依从性受多种因素影响。
关于礼来
礼来制药是一家致力于通过科学创新改善人类健康水平,惠及全球患者的医药公司。作为医疗健康行业的领军者,礼来制药拥有近150年的历史。今天,我们的药物已帮助全球超5100万人。
运用生物技术、化学和基因医学的力量,其科学家正在积极推动新的医学进展,以应对严峻的全球健康挑战。重新定义糖尿病与肥胖疗法,减少肥胖对人体的长期影响;助力阿尔茨海默病的防治行动;为一系列威胁人类健康的免疫性疾病提供解决方案;以及将难以治愈的癌症转变为可控的疾病。礼来制药迈向健康世界的每一步,都源自于其“致力于让数百万患者生活得更美好”的信念。这包括致力于解决全球多重挑战的创新临床试验,同时确保药物的可及性和可负担性。
参考文献
1.In a first-of-its-kind fixed dose study, once weekly insulin efsitora alfa leads to A1C reduction similar to daily insulin.
2.A Study of Insulin Efsitora Alfa (LY3209590) Compared to Glargine in Adult Participants With Type 2 Diabetes Who Are Starting Basal Insulin for the First Time (QWINT-1) (QWINT-1).
3.A Study of Insulin Efsitora Alfa (LY3209590) Compared With Insulin Degludec in Participants With Type 2 Diabetes Currently Treated With Basal Insulin.
4.中华医学会糖尿病学分会, 国家基层糖尿病防治管理办公室. 国家基层糖尿病防治管理手册(2022)[J]. 中华内科杂志, 2022, 61(7): 717-748.
5.国家老年医学中心, 中华医学会老年医学分会, 中国老年保健协会糖尿病专业委员会. 中国老年糖尿病诊疗指南(2024 版)[J]. 中华糖尿病杂志, 2024,16(2):147-189.
展开更多



