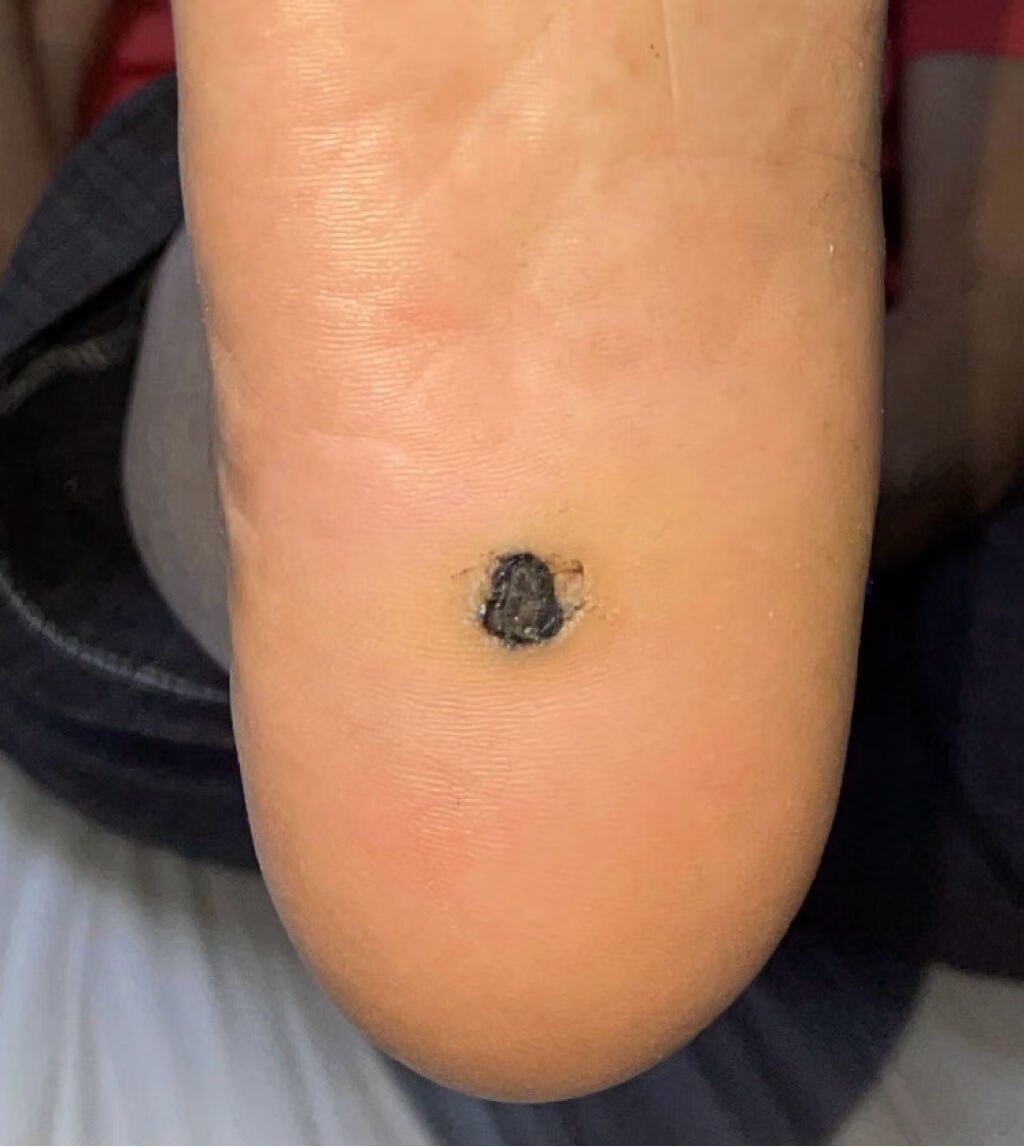
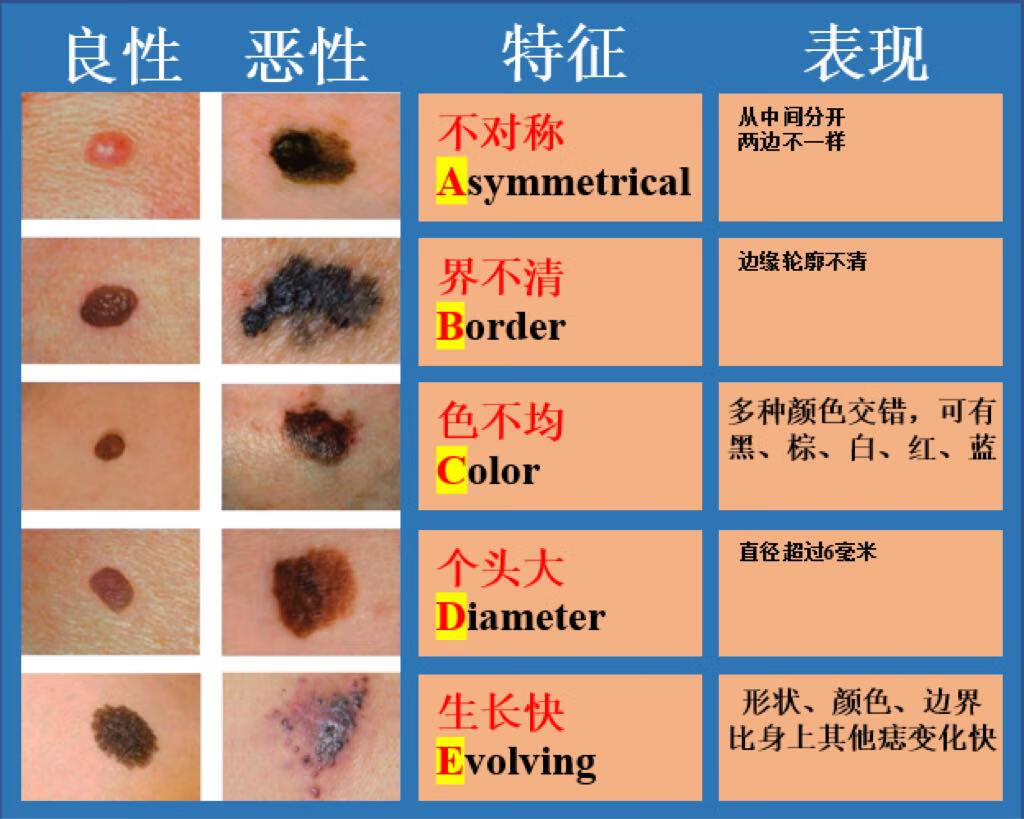
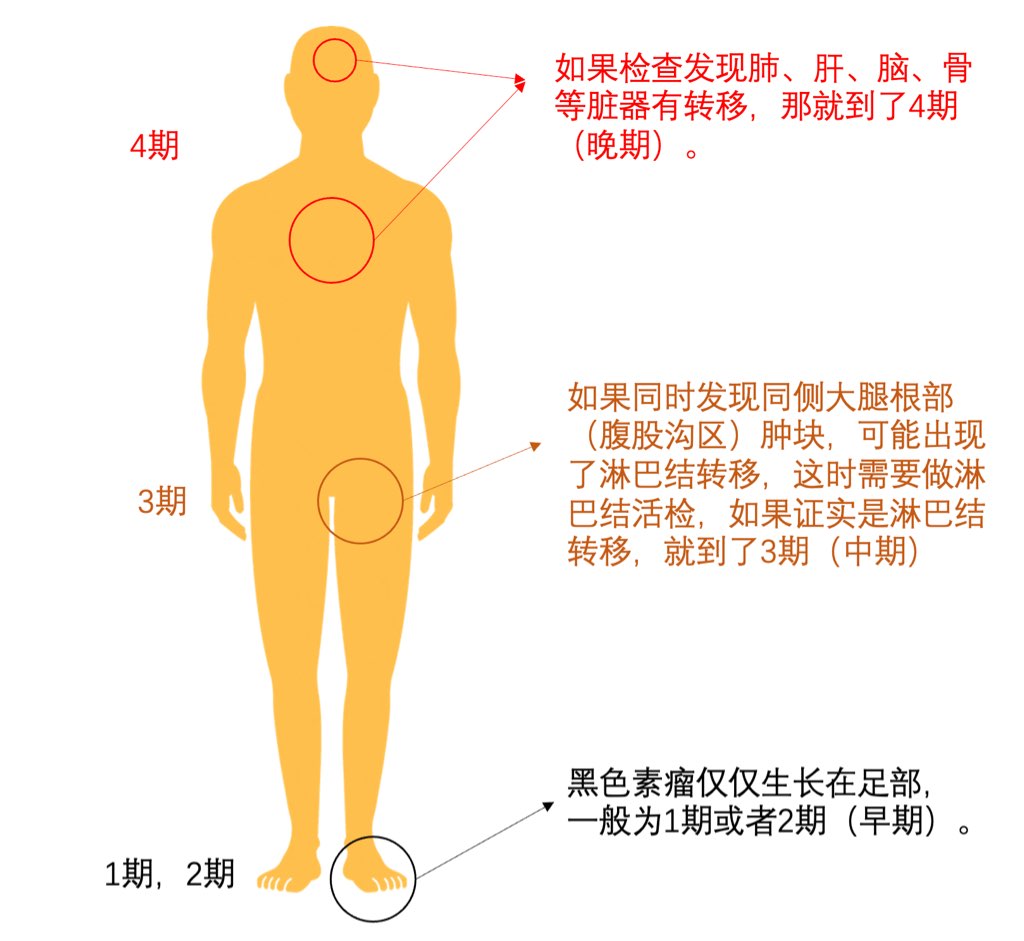
首都医科大学附属北京友谊医院始建于1952年,原名为北京苏联红十字医院,是新中国成立后,由党和政府建立的第一所大型综合性医院。建院初期,毛泽东、周恩来、刘少奇、朱德等老一辈革命家为医院亲笔题词。毛泽东主席特别题词“减少人民的疾病,提高人民的健康水平”。1957年3月,苏联政府将医院正式移交我国政府,周恩来总理来院参加了移交仪式。1970年,周总理亲自为医院命名为“北京友谊医院”。 目前,首都医科大学附属北京友谊医院已发展为集医疗、教学、科研、预防和保健为一体的北京市属三级甲等综合医院,是首都医科大学第二临床医学院。医院设有西城院区和通州院区,其中西城院区位于首都核心区,通州院区位于城市副中心。 北京友谊医院建设规模31.07万平方米,其中西城院区建设规模19.4万平方米,通州院区一期建设规模11.13万平方米。医院现有职工4400人,其中研究生导师150人,高级专业技术人员623人,国家级和北京市级专业委员会主委、副主委及核心期刊主编、副主编84人。目前两院区共开放床位2300张,年门诊量336万人次,年出院患者9.3万人次。北京友谊医院是北京市首批基本医疗保险A类定点医疗机构,可实现住院患者全国异地医保持卡结算,也是全国最早承担干部保健及外宾医疗任务的医院之一。 医院综合优势明显,专业特色突出,共有临床医技科室54个。消化和泌尿系统疾病诊治,肝、肾移植,肾内血液净化,热带病和寄生虫诊治以及中西医结合是医院的专业特色。2014年10月,医院获批成为国家消化系统疾病临床医学研究中心,2018年牵头成立北京市医院管理中心消化内科学科协同发展中心。 近年来,医院的医学科技创新能力显著提升,学科架构日臻完善,支撑平台不断强化,综合优势逐渐凸显。医院拥有国家临床重点专科8个,博士点27个,硕士点31个,国家住院医师规范化培训专业基地17个,国家专科医师规范化培训试点基地4个,“扬帆”重点专业7个,北京市重点实验室4个,北京市研究所4个,医学转化中心1个,还拥有支撑临床研究发展的国际标准化临床研究质控平台、ISO9001认证生物样本库和多中心互认医学伦理平台与研究型病房。医院与海外院校长期保持学术交流合作,接待国外专家学者短期交流以及留学生来院参观见习。自2005年起,北京市李桓英医学基金会已资助北京市14批次共231名中青年科技人才出国前往世界一流科研院所学习深造。 2012年7月1日,北京友谊医院作为全国和北京市医药卫生改革综合试点单位,率先实现“两个分开、三个机制”的改革试点。2016年4月,受北京市政府和平谷区卫计委的委托,北京友谊医院对平谷区医院以“区办市管”为模式进行管理。2019年7月,北京友谊医院顺义院区主体建设项目开工建设。同年12月,通州院区二期工程开工建设。医院先后于2017年4月8日和2019年6月15日启动了医药分开综合改革和医耗联动综合改革,坚持“医疗改革与提升医疗技术质量相结合,与改善患者就医感受相结合”。 多年来,北京友谊医院党委坚持以习近平新时代中国特色社会主义思想为指导,带领全院干部职工在推进医疗改革、改善医疗服务、提升医疗质量、创新驱动发展、落实非首都功能疏解和京津冀协同发展等方面,大胆改革,锐意进取,扎实工作,整体社会评价在全市及全国医院中名列前茅。在2018年度全国三级公立医院绩效考核中评价等级A+,北京友谊医院在参评的全国2398家公立医院中排名第19。在北京市医疗服务能力管理综合排名和北京市属三甲医院绩效考核中,北京友谊医院连续多年位居前三甲,医院消化内科、普外科在全市重点专科排名中位列第一。医院曾先后两次被授予全国先进基层党组织,曾获全国“三八”红旗集体、全国模范职工之家、中国质量奖提名奖、首都劳动奖章等荣誉,多次被授予首都文明单位标兵等光荣称号。 建院以来,北京友谊医院得到了党和国家领导人以及各级党委政府的关怀。在市委市政府、市卫生健康委和市医院管理中心的领导下,医院坚持“全心全意为患者服务”的宗旨,弘扬“仁爱博精”的院训精神,建立现代医院管理制度,坚持党委领导下的院长负责制,努力实现患者信任,职工幸福,医院发展,党和政府放心。未来,医院将以国家消化学科群为战略学科,整合现有国家临床重点专科项目、传统特色学科、有发展潜能的优势学科,发挥医院综合实力,创新驱动发展,努力把医院建设成为国家级医学中心,形成职工共同追求的友谊梦,为首都医药卫生事业的发展做出新的更大的贡献。珠蛋白生成障碍性贫血原名地中海贫血又称海洋性贫血,是一组遗传性溶血性贫血疾病。由于遗传的基因缺陷致使血红蛋白中一种或一种以上珠蛋白链合成缺如或不足所导致的贫血或病理状态。缘于基因缺陷的复杂性与多样性,使缺乏的珠蛋白链类型、数量及临床症状变异性较大。根据所缺乏的珠蛋白链种类及缺乏程度予以命名和分类。 本病广泛分布于世界许多地区,东南亚即为高发区之一。我国广东、广西、四川多见,长江以南各省区有散发病例,北方则少见。,1.β珠蛋白生成障碍性贫血(β地中海贫血) β珠蛋白生成障碍性贫血(简称β地贫)的发生的分子病理相当复杂,已知有100种以上的β基因突变,主要是由于基因的点突变,少数为基因缺失。 2.α珠蛋白生成障碍性贫血(α地中海贫血) 大多数α珠蛋白生成障碍性贫血(地中海贫血)(简称α地贫)是由于α珠蛋白基因的缺失所致,少数由基因点突变造成。白基因的缺失所致,少数由基因点突变造成。,血液,0,缺铁性贫血、传染性肝炎或肝硬化,无,根据临床特点和实验室检查,结合阳性家族史,一般可作出诊断。有条件时可作基因诊断。 对于少见类型和各种类型重叠所致的复合体则非常复杂,临床表现各异,仅根据临床特点和常规实验室血液学检查是无法诊断的。而且由于基因调控水平的差异,相同基因突变类型的患者不一定有相同的临床表现。血红蛋白电泳检查是诊断本病的必备条件,但输血治疗后的血液学检查会与实际结果有所不同。所以进行遗传学和分子生物学检查才能最后确诊。遗传学检查可确定为纯合子、杂合子以及双重杂合子等。,。












 7
7
 104
104
 3
3
 1
1
 0
0
 1
1
 1
1
 1
1
 1
1
 4
4