简称:

住院医师
好评率:100%
立即咨询
-
*婧回复质量:非常满意服务态度:暂无回复速度:暂无functions/format.html图文问诊
-
*阳阳回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
-
*文凤回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
-
*名用户回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
-
*名用户回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
-
匿名用户回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
-
匿名用户回复质量:暂无服务态度:暂无回复速度:暂无functions/format.html此用户未填写评价内容图文问诊
展开更多
-
总交流次数
39
总回复次数
25
患者:男 16岁 -
总交流次数
18
总回复次数
11
患者:男 22岁 -
总交流次数
67
总回复次数
38
患者:女 2岁3个月 -
总交流次数
11
总回复次数
8
患者:女 -
总交流次数
36
总回复次数
22
患者:男 21岁 -
总交流次数
37
总回复次数
17
患者:女 -
总交流次数
22
总回复次数
11
患者:男 57岁 -
总交流次数
15
总回复次数
10
患者:女 -
总交流次数
7
总回复次数
6
患者:女 -
总交流次数
9
总回复次数
7
患者:女
展开更多
-

中国研究型医院学会神经科学专业委员会, 中国出生缺陷干预救助基金会神经与肌肉疾病防控专项基金组织专家组. 脊髓性肌萎缩症新生儿筛查专家共识(2023版) [J] . 中华医学杂志, 2023, 103(27) : 2075-2081. DOI: 10.3760/cma.j.cn112137-20230310-00372.
摘要脊髓性肌萎缩症(SMA)是一种常见的退行性神经肌肉疾病,表现为进行性肌肉无力和萎缩。近年来,相继出现的疾病修正治疗药物改变了SMA的自然病程,症状前诊断并给予疾病修正治疗优于症状后治疗。为此,本共识组织全国相关领域专家,围绕以下主题达成共识:SMA新生儿筛查流程与相关问题、SMA筛查后确诊流程与相关问题、筛查确诊SMA新生儿的疾病管理等,以期规范和指导当前SMA的新生儿筛查工作。脊髓性肌萎缩症(SMA)是一种由于脊髓前角α-运动神经元退化变性,导致肢体和躯干进行性、对称性肌无力和肌萎缩的常染色体隐性遗传性疾病,常伴有呼吸、消化和骨骼等多系统损害[1]。临床上以运动神经元存活基因1(SMN1)缺失和(或)微小变异导致的5q SMA类型最为常见。新生儿的SMA发病率约为1/10 000[1];中国人群SMN1基因7号外显子缺失携带率为1/42~1/48[2]。SMA具有高致残和致死率。已上市的三款SMA疾病修正药物(诺西那生钠、利司扑兰和索伐瑞韦)可显著改善疾病预后,改变疾病自然病程。SMA的新生儿于症状前诊断并给予疾病修正治疗,优于症状后治疗[3]。目前大多数欧美国家及日本、韩国等亚洲国家均已开展全国范围的SMA新生儿筛查[4-6]。近年来,中国大陆和台湾地区也有多个SMA新生儿筛查研究[7-8]。2021年《中国新生儿筛查专家共识:高通量测序在单基因病筛查中的应用》[9]中也将SMA纳入建议筛查疾病列表。SMA的新生儿筛查工作将在国内广泛开展与推广,此时迫切需要相关领域的指南来指导、规范相关工作。在此背景下,中国研究型医学学会神经科学专业委员会联合中国出生缺陷干预救助基金会神经与肌肉疾病防控专项基金组织专家组,开展本共识的撰写。一、本共识制订的方法学本共识以“脊髓性肌萎缩症、新生儿筛查、遗传学诊断、治疗、运动神经元基因、SMN基因、实践指南、专家共识、研究进展、多学科、神经肌肉疾病、疫苗;newborn screening、neonatal screening、spinal muscular atrophy、dried blood spot screening、rare diseases、public health、newborn bloodspot screening、epidemiology、birth prevalence、neuromuscular disease”等为检索词,以Pubmed、web of science、EMBASE、中国知网、万方医学、中国生物医学文献数据库等数据库为检索范围,检索2000年1月至2023年3月发表的相关国内外SMA新生儿筛查实践和国际指南共识为参照,由新生儿筛查学、医学遗传学及临床医学等领域的29位专家反复讨论制订,并发放给全国不同等级医院相关专业医生,提出意见并综合整理修改,旨在为实验室人员和医生的临床诊疗提供指导和帮助。本共识以“问题,推荐意见”为框架对每一条推荐意见进行投票。对有争议的推荐意见多次讨论,循证共识,最终得出推荐意见。二、SMA新生儿筛查流程与相关问题1. SMA新生儿筛查的目标疾病与基因:研究证明95%的SMA患者存在SMN1基因7号外显子的纯合缺失[10],且基因型-表型关联明确。因此针对SMN1基因7号外显子纯合缺失的筛查方法可靠,适宜在全体新生儿范围内进行推广。未累及SMN1基因7号外显子纯合缺失的其他SMN1变异所致的SMA,因缺乏热点变异位点且不能被常用的SMN1基因7号外显子拷贝数定量方法检出,尚不具备纳入新生儿筛查的条件。美国新生儿统一筛查推荐疾病列表(RUSP)也明确将SMA列为“由SMN1基因7号外显子纯合缺失所致的疾病”[10]。因此,现阶段SMA新生儿筛查的目标疾病仅包括累及SMN1基因7号外显子纯合缺失所致的SMA。由SMN1基因的其他类型突变所致的SMA及非SMN1基因突变所致的SMA尚未纳入SMA新生儿筛查的目标疾病,此类SMA患者尚无法通过目前采用的新生儿筛查方法被检出,应在知情同意中明确告知。2. SMA新生儿筛查的方法及其筛查效能:SMN1基因7号外显子纯合缺失有多种不同检测方法可供选择。筛查一般采用针对7号外显子的定量检测方法,如荧光定量聚合酶链反应(PCR)、多重连接依赖的探针扩增技术(MLPA)、变性高效液相色谱(DHPLC)等。筛查机构应根据国家法规要求、行业技术规范和本机构实验室的实际情况等选择适宜的方法开展筛查。虽然通过定量技术判断SMN1基因7号外显子纯合缺失是可靠且成熟的方法,但是对于筛查而言,受限于样本质量等多重因素,仍无法完全排除假阳性和假阴性的可能。综合我国国家药品监督管理局已注册产品的性能指标、临床常用方法学技术特征和国内外新生儿筛查研究报告数据等,筛查机构选择的筛查方法对于SMN1基因7号外显子纯合缺失的检测敏感性应≥95%,阳性预测值应≥90%[4-6,11-14]。SMN2基因拷贝数是SMA的重要修饰因素,但并非筛查检测所必须,并且可能会增加筛查成本。因此,临床开展SMA新生儿筛查可以不包括定量检测SMN2基因拷贝数。但SMN2基因拷贝数对确诊SMA新生儿的后续临床治疗有重要指导价值,因此筛查阳性的新生儿的确诊试验应包含SMN2基因拷贝数信息。3. SMA新生儿筛查的操作流程:新生儿筛查的程序包括健康教育与知情同意、标本采集与递送、实验室检测、阳性病例确诊、分型及治疗与随访等众多环节,具体程序可见筛查、确诊及疾病管理流程图(图1)。SMA新生儿筛查实验室所在医疗机构、技术人员和仪器试剂的资质以及实验室工作区域及仪器配置、数据解读和质量控制等方面均应符合我国《新生儿疾病筛查技术规范(2010版)》和《医疗机构临床基因扩增检验实验室管理办法》的相关规定要求[15-16]。SMA新生儿筛查为基因筛查,基本不受孕周、出生时间、蛋白负荷、进食及药物等因素的影响,对血片采集时间亦无特定要求,但建议与我国目前现行的新生儿筛查同时采集或尽早采集。不含白细胞成分的输血治疗不影响样本采集(全血输注者应在3个月后采集);对于拟行骨髓移植或干细胞移植的新生儿应在治疗前完成采集。为避免因DNA质量影响实验室检测效率,建议用于检测的滤纸干血斑直径应≥6 mm。注:SMA为脊髓性肌萎缩症;SMN2为运动神经元存活基因2;SMN1为运动神经元存活基因1图1 新生儿脊髓性肌萎缩症(SMA)筛查、确诊及疾病管理流程图4. SMA新生儿筛查的质量控制:筛查实验室应建立并重视SMA新生儿筛查的质量控制。应采用与采集样本(滤纸干血片)相匹配的DNA提取试剂盒,保证样本DNA能够达到所选用的SMN1基因检测试剂盒的要求。应建立新生儿SMA筛查检测的质量管理体系:(1)对采用的检测系统的性能进行验证或确认,评估结果与选用的试剂盒操作说明书一致,方可用于临床服务;(2)实验室应建立适当的室内质量控制规则以监控系统误差和随机误差,做好质量控制记录并定期分析,持续改进;(3)根据所选用检测试剂盒性能指标与本地检测数据,确定SMN1基因7号外显子纯合缺失筛查的阳性切值,以保证检测敏感性和阳性预测值符合本专家共识的建议。专家组建议在筛查实践中开展系统性研究,推动建立SMN1基因7号外显子纯合缺失全国性的室间质量评价标准。推荐意见1:目前SMA筛查仅针对SMN1基因7号外显子纯合缺失类型(同意率100%);筛查机构选择的筛查方法对于SMN1基因7号外显子纯合缺失的检测敏感性应≥95%,阳性预测值应≥90%(同意率100%);SMA新生儿筛查可以不包括定量检测SMN2基因拷贝数,但筛查阳性新生儿的确诊试验应包含SMN2基因拷贝数信息(同意率100%);用于检测的滤纸干血斑直径应≥6 mm,全血输注者应在3个月后采集血标本,对于拟行骨髓移植或干细胞移植的新生儿应在治疗前完成采集(同意率100%)。三、SMA筛查后确诊流程与相关问题1. 筛查阳性后的家系召回和咨询:发现标本筛查阳性的新生儿,应立即召回,同时采集核心家系成员全血进行确诊性基因检测。确诊实验应包括SMN1及SMN2拷贝数检测,同时对患儿进行详细的体格检查,并行运动功能及电生理等评估,以便尽早确诊,依据SMN2拷贝数并结合临床评估以决策治疗。2. 筛查假阳性的原因:新生儿SMA筛查假阳性的概率极低,其大多由筛查流程缺少规范化与检测技术本身的局限性所造成[17]。如:新生儿筛查滤纸干血片质量不高(血量少,血斑污染,血片高温、高湿环境保存或血片递送不当等)、筛查实验室方法的选择及临界值的确定不准确、实验室技术人员操作欠规范和新生儿白细胞计数降低等因素均可引起假阳性[18]。3. 与筛查阳性的新生儿监护人沟通与咨询的内容:与筛查阳性的新生儿监护人进行充分沟通,并提供疾病相关的咨询,对SMA新生儿筛查及后期疾病管理至关重要。建议沟通与咨询应包括以下内容:(1)疾病相关信息、筛查结果及其代表的意义;(2)筛查技术存在的局限性和目前结果存在的各种可能性;(3)立即到指定的具备诊断资质医疗机构,采集新生儿及核心家系成员血样本(新生儿和生物学父母)进行确诊实验,神经肌肉病专家同步检查和评估新生儿临床指标;(4)告知确诊实验所采用的技术、风险及局限性;(5)明确告知:如确诊试验为阴性,仍需定期随访并纳入儿童保健系统管理;如确诊为阳性,立即参与由儿童神经病学和医学遗传学等医生组成的医疗团队的治疗讨论,选择个体化诊疗方案,建议监护人积极接受并参与由政府、社会组织等(特殊疾病救助渠道、病患组织、病友群等)提供的各种帮助,树立长期随访的准备和信心[19];(6)在确诊的等待期,家长(监护人)需要密切关注新生儿运动、喂养和呼吸等方面的表现,一旦出现异常表现(如哭声变弱、喂养困难、已获得的运动功能减退或丧失、运动能力落后于年龄、腹式呼吸和生长障碍等),则需立即就医[20-21]。遗传咨询首先应获取监护人的信任,以通俗易懂的语言解释问题,真诚温暖的方式缓解和抚慰监护人焦虑情绪[22]。4. 对召回的阳性新生儿及其核心家系成员的确诊实验:对召回的阳性新生儿及其核心家系成员应立即进行确诊实验。根据国内外的SMA遗传学诊断共识[23-24],确诊实验推荐定量分析的技术,如国际公认的金标准技术MLPA、液滴数字PCR(ddPCR)、荧光定量PCR技术等[19]。召回新生儿的确诊实验需同时包括SMN2拷贝数的检测,有助于SMA分型预判、症状前治疗策略的制定及后期随访规划。核心家系成员SMN1基因型等遗传传递信息亦需检测,为进一步的遗传咨询提供支持。1型SMA的平均发病年龄是2~3个月[25-26],最严重的1a型通常<1个月。为了能够及时开展症状前治疗,确诊实验建议在2周内完成,应依据《脊髓性肌萎缩症遗传学诊断专家共识》[24]出具规范化的基因诊断报告。根据国内外的SMA新生儿筛查实践经验,自出生进行新生儿筛查再到确诊实验的完成,周期为15~30 d[17-18,27]。确诊实验需警惕标本污染导致的假阴性,要确保标本留取、运输及实验的规范化与准确性。同时要警惕假阳性,譬如位于探针杂交区域或者引物结合位置的变异会导致杂交或者扩增失败而出现假阳性[17],但是这些情况发生的概率较低。5. 确诊实验完成后与家属沟通的内容:确诊实验完成后,需再次与召回的新生儿筛查阳性家庭进行沟通,提供咨询和指导。建议沟通内容主要包括:(1)确诊实验结果为阳性,应立即联系医学遗传学专家和儿童神经病学专家团队报告确诊实验的结果并进行讨论,由上述专家团队对讨论的结果进行解释,告知疾病相关信息、家系遗传传递规律和再发风险评估;同时建议家系其他成员进行扩展性筛查,以期早期明确其他成员是否患病及存在的风险。由SMA专科诊治医生为确诊阳性患儿进行认知、语言、运动功能、喂养、呼吸和睡眠等的评估,依据临床评估与SMN2拷贝数制定个体化治疗策略,同时提供后期多学科的管理和日常护理指导。(2)确诊实验结果为阴性,同样需要及时联系医学遗传学专家和儿童神经病学专家报告实验结果,由遗传专家或儿科神经学专家对筛查的家庭进行遗传咨询和指导:解释筛查阳性而确诊阴性的原因及可能存在的复合杂合突变类型SMA等残余风险(风险约为5%);同时建议针对这些家庭制定随访计划,每3个月随访一次,至2岁结束,指导家长密切关注孩子的认知、语言、运动功能、呼吸、消化和睡眠等的发育与变化,及时发现异常情况,及早就诊获得医疗帮助和支持。推荐意见2:发现筛查阳性的标本,应立即召回对应的新生儿,依据SMN2拷贝数结合临床评估决策治疗(同意率100%);确诊实验推荐定量分析的技术,如国际公认的金标准技术MLPA、ddPCR、荧光定量PCR技术等(同意率100%);确诊实验建议应在2周内完成,自出生进行新生儿筛查到确诊实验的完成,建议周期为15~30 d(同意率100%)。四、筛查确诊阳性后SMA新生儿疾病管理1. 筛查确诊后SMA新生儿治疗策略选择:经新生儿筛查确诊阳性的(有/无症状)SMA患儿均需要及时制定个体化治疗与疾病管理方案。药物治疗选择,需根据国内已获批药物的适应症进行合理临床应用。多个国家的SMA新生儿筛查治疗专家共识或相关临床研究建议将SMN2拷贝数作为是否启动症状前治疗的生物标志物[28-31]。SMN2拷贝数为1且已出现临床症状的SMA患儿,依据家长意愿酌情考虑是否启动治疗;SMN2拷贝数为1的无症状的SMA患儿及SMN2拷贝数为2(临床分型最可能为1或2型)或拷贝数为3(临床分型最可能为2或3型)的无症状SMA患儿,均建议立即启动治疗;SMN2拷贝数为4的无症状SMA患儿(预测未来临床分型为3或4型),建议尽早启动疾病修正治疗[32-33];SMN2拷贝数为5的无症状SMA患儿建议严密观察及随访,暂不立即启动治疗[32](图1)。2. 筛查确诊后SMA新生儿的随访管理:新生儿筛查确诊的SMA患儿需接受长期、规范的医学随访。SMN2拷贝数为1~4的无症状SMA患儿在接受治疗后应每3个月完成1次医学随访[12]。因各种原因未能及时接受治疗的无症状SMA患儿,6月龄内的建议每月随访1次;6月龄至2岁的建议每2~3个月随访1次;2岁以上的每6~12个月随访1次。常规随访内容建议包括尺神经或腓神经复合肌肉动作电位(CMAP)波幅等电生理检查、神经系统体格检查以及运动量表费城儿童医院婴儿神经肌肉性疾病测试(CHOP INTEND)、Hammersmith婴儿神经系统检查(HINE)、Hammersmith运动功能量表扩展版(HEMSE)等相关评估[12,28]。未接受疾病修正治疗的无症状患儿,随访过程中需密切监测SMA的发病预警信号,一旦患儿出现吞咽能力或呼吸模式的显著变化、已获得的运动能力出现下降或预期运动能力未能获得以及CMAP波幅低于同年龄正常值等相关征象,建议立即启动疾病修正治疗。对症状性SMA患儿,推荐成立多学科联合门诊,针对不同功能状态的患者,制定个体化诊疗建议,包括神经、康复、骨科、呼吸、消化、营养及护理等在内的多学科管理应贯穿于SMA疾病治疗的全过程[34-35]。推荐意见3:SMN2拷贝数为1且已出现临床症状的SMA患儿,依据家长意愿酌情考虑是否启动治疗(同意率100%);SMN2拷贝数为1的无症状的SMA患儿及SMN2拷贝数为2或3的无症状SMA患儿均建议立即启动治疗(同意率100%);SMN2拷贝数为4的无症状SMA患儿,建议尽早启动疾病修正治疗(同意率100%);SMN2拷贝数为5的无症状SMA患儿建议严密观察随访,暂不立即启动治疗(同意率100%)。推荐意见4:SMN2拷贝数为1~4的无症状SMA患儿在接受治疗后应每3个月完成1次医学随访(同意率100%)。因各种原因未能及时接受治疗的无症状SMA患儿,6月龄内的建议每月随访1次;6月龄~2岁的建议每2~3个月随访1次;2岁以上的每6~12个月随访1次(同意率100%)。五、筛查确诊后SMA新生儿疫苗接种问题对于经新生儿筛查确诊后的无症状的SMA患儿可按我国儿童疫苗接种程序接种,必要时进行疫苗接种相关评估,尤其需特别关注并及时接种肺炎疫苗或流感疫苗等呼吸道疾病相关的疫苗[36]。尽管目前尚无SMA患者接种疫苗与疾病修正治疗药物疗效或及安全性相关的研究报道,但不建议在免疫接种后72 h内进行疾病修正治疗;有建议接种7 d后无不良反应发生者,可进行诺西那生钠鞘内注射治疗[37-38]。推荐意见5:无症状的SMA患儿可按我国儿童疫苗接种程序接种(同意率100%);不建议在免疫接种后72 h内进行疾病修正治疗(同意率100%)。SMA疾病修正治疗药物的症状前应用可显著改善患儿预后,而新生儿筛查则是发现症状前SMA患者的最佳途径。因此,开展SMA新生儿筛查已成为当前全球新生儿疾病筛查的热点。SMA疾病修正治疗药物诺西那生和利司扑兰分别于2022年和2023年被纳入国家医保目录,使得在我国开展SMA新生儿筛查的条件充分成熟。在知情同意下,严格遵守操作流程与质量控制,筛查由SMN1基因7号外显子纯合缺失所致的SMA患儿,技术成熟,路径方便可行。确诊后,依据SMN2拷贝数及临床症状,选择酌情或及时或立即给予药物治疗,对所有患者进行定期、规范化、多学科综合随访管理,使患者得到最大获益。SMA新生儿筛查工作不仅关系到技术,也是公共卫生政策的组成部分。在我国,SMA筛查的实施还面临疾病知晓率低、筛查费用支付高、治疗与康复普及困难等多方面挑战。因此,SMA新生儿筛查应在知情同意与自主选择的原则下,在有条件的单位和地区逐步开展。本共识仅代表参与编写讨论专家的观点,不具备法律效力。共识发布后,中国研究型医院学会神经科学专业委员会将随时关注相关领域的证据变化情况,按照共识方法学要求,每年回顾相关推荐意见的证据变化,根据变化情况组织本领域专家对推荐意见进行相应修订,一般情况下每3~5年对共识进行一次全面修订。参考文献:略 -

一 mtDNA的结构特点
mtDNA是指位于线粒体中的DNA,而真核细胞中大部分DNA存在于细胞核中。线粒体是动物细胞核外唯一含有DNA的细胞器,它存在于除成熟红细胞外的所有组织细胞中。人类mtDNA是一个全长16569bp的双链闭合环状分子,外环为重(H)链,内环为轻(L)链。人类mtDNA包含37种编码基因,分别编码13种多肽链、22种tRNA和2种rRNA。线粒体上的基因编码紧凑,没有内含子。mtDNA唯一的非编码区是约1000bp的D-环,它包含mtDNA重链复制起始点、轻重链转录的启动子以及4个高度保守的序列,分别位于213-235、299-315、346-363以及终止区16147-16172。mtDNA两条链的碱基组成差别较大,H链富含G,而L链多含C。H链是12种多肽链、2种rRNA和14种tRNA转录的模板,而L链仅作为1种多肽链和8种tRNA转录的模板。mtDNA具有两个复制起始点,分别起始复制H链和L链,其H链复制的起始点(OH)与L链复制起始点(OL)相隔2-3个mtDNA。与核DNA不同,mtDNA分子上无核苷酸结合蛋白,缺少组蛋白的保护,基因与基因之间少有间隔,而且线粒体内无DNA损伤修复系统,这些因素成为mtDNA易于突变且突变难以修复并遗传到子代细胞的分子基础。mtDNA的另一特点是每一个细胞中含有数百个线粒体,每个线粒体内含有2-10个拷贝的mtDNA分子,每个细胞可具有数千个mtDNA分子,从而构成细胞中mtDNA异质性的分子基础。线粒体结构图二 mtDNA的遗传特征与核DNA相比,mtDNA具有独特的遗传规律。了解线粒体的遗传规律可以更好地认识线粒体疾病的病因学与发病机制。1、mtDNA具有半自主性线粒体具有自己的遗传物质,所以有人将mtDNA称为第25号染色体或M染色体。mtDNA能够独立地复制、转录和翻译,但是大量的维持线粒体结构和功能的大分子复合物,以及大多数氧化磷酸化酶的蛋白质亚单位均由核DNA编码,故mtDNA的功能又受核DNA的影响,因而是一种半自主复制体。2、线粒体基因组所用的遗传密码和通用密码不同线粒体的遗传密码和通用密码并不完全一样,核基因组中UGA为终止密码子,而在mtDNA中UGA编码色氨酸。另外,线粒体的tRNA兼并性较强,仅用22个tRNA就可识别多达48个密码子。3、mtDNA为母系遗传人类受精卵中的线粒体来自于卵细胞,也就是说来自母系,这种传递方式称为母系遗传。这是因为卵细胞含有十多万个mtDNA分子,而精子只有大约几百个,相对于卵子而言,精子对线粒体基因型的影响很小。另一原因是用于推动精子运动的大量线粒体存在于精子底部,在受精时精子尾部会丢失,从而导致精子中的mtDNA不能进入卵细胞。由于受精过程和细胞分裂过程中线粒体与细胞核的行为不同,导致线粒体遗传病的传递模式不同于经典的孟德尔遗传。因此,如果在某个家族中发现一些成员具有相同的临床症状,并且是从受累的女性传递下来,就应考虑线粒体DNA突变的可能性。4、mtDNA在细胞分裂过程中的复制分离与遗传瓶颈现象线粒体DNA在减数分裂和有丝分裂期间都要经过复制分离。人类卵母细胞中虽含有约十多万个线粒体,但在卵母细胞成熟中绝大多数线粒体会丧失,数目减至10-100个。这种卵细胞成熟过程中线粒体数目从十万个锐减到少于100个的过程就是遗传瓶颈,使得只有少数线粒体真正传给后代,也是造成亲代与子代之间差异的原因。此后,经过早期胚胎细胞分裂,线粒体通过自我复制使数目达到每个细胞含有十万多个或更多。如果通过遗传瓶颈保留下来的一个线粒体碰巧携带一种突变基因,那么这个突变基因就可能在发育完成之后的个体中占有一定的比例。由于在胚胎发生和组织形成的细胞分裂过程中线粒体经过复制分离,随机进入子细胞。因此,一些子细胞很可能接受大量的携带突变基因的线粒体,由他们形成的成体组织细胞会具有较高比例携带突变基因的线粒体。5、mtDNA的异质性与阈值效应如前所述,人类每个细胞中都有数千个乃至十万个mtDNA分子。纯质是指细胞或组织中所有的线粒体具有相同的基因组,即都是野生型序列,或者都是突变型序列。异质则表示一个细胞或组织既含有野生型,又含有突变型线粒体基因组。在异质性细胞中,突变型与野生型mtDNA的比例决定了细胞是否出现能量短缺。如果携带突变型线粒体比例较小,则产能不会受到明显影响。相反,当含有大量突变型线粒体基因组的组织细胞所产生的能量不足以维持细胞的正常功能时,就会造成组织中能量供应水平降低,进而影响组织的功能并出现异常的性状。也就是说,当突变的mtDNA达到一定比例时,才有受损的表型出现,这就是阈值效应。这种线粒体基因突变产生有害影响的阈值效应明显依赖于受累细胞或组织对能量的需求。因此,高需能的组织,如脑、骨骼肌、心脏和肝脏等,更容易受到mtDNA突变的影响。6、mtDNA的突变率极高mtDNA的突变率比核DNA高10-20倍。这种高突变率造成个体及群体中mtDNA序列差异较大。比较任何两个人的mtDNA,平均每1000个碱基对中就有4个不同。人群中含有多种中性到中度有害的mtDNA突变,且高度有害的mtDNA突变也会不断增多。不过有害的突变会由于选择而被消除,故突变的mtDNA基因虽然很普遍,但线粒体遗传病却不常见。三、线粒体基因突变的类型1、碱基突变(1)错义突变:通常发生于mtDNA中的蛋白质编码序列上,导致所编码的氨基酸发生改变。主要与脑脊髓性及神经性疾病有关,如Leber遗传性视神经病和神经肌病等。(2)蛋白质生物合成基因突变,多为tRNA基因突变。与错义突变引起的疾病相比,这类突变所致的疾病更具有系统性的临床特征,而且几乎所有突变都是tRNA突变,并与线粒体肌病相关。典型的肌病包括肌阵挛性癫痫伴碎红纤维病综合征、线粒体肌病脑病伴乳酸酸中毒及中风样发作综合征、母系遗传的肌病及心肌病等。2、缺失、插入突变以缺失突变更多见。这类疾病往往无家族史,散发。导致mtDNA缺失的原因多为mtDNA的异常重组或在复制过程中的异常滑动。常见于神经性疾病及一些退化性疾病,如KSS综合征。绝大多数的眼肌病是由缺失突变引起。3、mtDNA拷贝数目突变拷贝数目突变指mtDNA拷贝数大大低于正常,这种突变较少,仅见于一些致死性婴儿呼吸障碍、乳酸中毒或肌肉病变及肝、肾衰竭的病例。此外,mtDNA病变还具有相应的组织特异性。不同组织对氧化磷酸化依赖性的差异是线粒体病组织特异性的基础。有人认为,这种依赖性的差异是由核DNA编码的氧化磷酸化基因的组织特异性调控造成的。应注意的是,氧化磷酸化过程中5种酶复合物是由mtDNA和核DNA共同编码,编码这些酶的核基因突变也可能产生类似于线粒体病的症状。因此,有些线粒体遗传病是核DNA与mtDNA共同作用的结果。四、常见线粒体遗传病作为细胞的能量代谢中心,线粒体一旦出现功能改变就会导致病理变化。随着对线粒体生物化学和遗传学认识的不算深入,所发现的线粒体遗传病也在逐渐增多。人类首先识别的线粒体疾病是Leber遗传性视神经病,其临床表现为在中年时突发失明。数十年的线粒体基因突变的积累则会导致生物个体衰老、退行性疾病和肿瘤的发生。由于线粒体是母系遗传,而且卵细胞线粒体的数目非常多,线粒体突变并非涉及所有的线粒体,这也是线粒体疾病复杂的病理表型的分子机制。在一个线粒体疾病家族中,由于突变型线粒体在线粒体总数中所占比例不同,家族成员的临床表型可以从正常表型到非常严重的综合征并存,并且患者的发病年龄也不尽相同。只有细胞中突变型线粒体达到一定比例,线粒体产生能量的能力下降到一定的阈值时,细胞才会丧失其正常的功能。高度依赖于氧化磷酸化的高需能组织器官,例如,神经系统和心脏,在mtDNA发生突变时遭受的损害更为严重。线粒体疾病种类很多,而且原因各不相同。一部分疾病完全是由于mtDNA的异常引起的,如Leber遗传性视神经病、MERRF综合征等;还有一部分疾病,发病原因可能部分与线粒体异常有关,如糖尿病、癌症及心肌疾病的产生、乳酸酸中毒、某些 肌病、骨质疏松症、阿尔兹海默症、帕金森病、中风等。此外,人类的衰老也与mtDNA突变有关。 -

基因组(Genome):分子生物学和遗传学领域中指生物体所有遗传物质的总和,包括DNA或RNA(病毒)。具体包含编码DNA、非编码DNA、线粒体DNA和叶绿体DNA。研究基因组的科学称为基因组学。
同源染色体(Homologous Chromosomes):一个物种中形态和结构基本相同的染色体。在二倍体生物细胞中,同源染色体在减数第一次分裂的四分体时期中彼此联会(若是三倍体及其他奇数倍体生物细胞,联会时会发生紊乱),最后分开到不同的生殖细胞中。子代的一对染色体其中的一条来自母方,另一条来自父方。参考基因组(Reference Genome, REF):又称参考(序列)组装(A Reference Assembly),是一个电子化的核酸序列数据库(A digital nucleic acid sequence database)。它由多个科学家和研究单位协作组装、维护和更新,用以作为一个物种的一个理想化的个体的、全基因组序列的典型代表或案例(但不能保证可以精准地代表某个地球上存在过的生物体)。人类、病毒、细菌、真菌、植物和动物理论上都有各自的参考基因组,目前只有部分物种被测通和公布。人类基因组由23对染色体、约60亿个碱基(或核苷酸)组成。正常人类基因组是以2个拷贝存在(是指同源染色体,而非姐妹染色单体),分别来自父母。人类的基因组有几个不同的版本名,目前比较常用的有hg19、hg38、GRCh37、GRCh38。hg系列是UCSC的叫法,GRCh系列是NCBI和Ensembl的叫法。同一版本的序列是一样的,hg19对应GRCh37,hg38对应GRCh38(坐标与hg19/GRCh37不同)。参考基因组的实体是一个文本文件(.fasta),通常是个单倍体(除了性染色体),含有染色体号和核酸(A/T/G/C)序列,可压缩与索引,且包含一系列的配套文件(例如:GTF文件,记录每个基因名称及其各种元器件的位置)。参考基因组可提供来自每个供体不同DNA序列的单倍体镶嵌(A haploid mosaic of different DNA sequences from each donor)。事实上,基因组学、高通量测序以及相关的生信分析技术,很大程度上得益于人类基因组计划(Human Genome Project, HGP)。HGP是一项与曼哈顿原子弹计划和阿波罗计划相提并论的规模宏大、跨国跨学科的科学探索工程。旨在测定组成人类染色体(指单倍体)中所包含的30亿个碱基对组成的核苷酸序列,从而绘制人类基因组图谱,并且辨识其载有的基因及其序列,达到破译人类遗传信息的最终目的。全基因组测序(Whole Genome Sequencing, WGS):是指利用高通量测序平台对生物的不同个体(或群体)、同一个体的不同器官(或组织、细胞)进行全基因组测序,并进行生物信息学分析(主要是利用统计方法获取影响表型或经济性状的候选基因或功能突变)。高通量测序(High-Throughput Sequencing, HTS):是对传统Sanger测序(也称为一代测序技术)革命性的改变, 一次(一轮反应或拍照)对几十万到几百万条核酸分子进行序列测定,故又称下一代测序技术(Next Generation Sequencing,NGS)。高通量测序也被称为深度测序(Deep Sequencing),是人类历史上多学科、基础研究、资本运作与商业化结合的成功案例之一,直接导致了高通量测序仪(当前世界最尖端的大型设备之一)的发明与革新,使得对一个物种的基因组和转录组进行高效、细致、全貌的分析成为常规操作。高通量测序作为分子群体遗传学和个人基因组学研究的有力工具,对21世纪前半叶的生命科学研究、生产、疾病的诊断和治疗起到巨大作用,也对生物信息学的进一步发展起到重要的推动作用。基因结构:真核生物的基因大致分为4个区域,1)编码区,包括外显子与内含子;2)前导区,位于编码区上游,相当于RNA 5’末端非编码区(非翻译区);3)尾部区,位于RNA 3’编码区下游,相当于末端非编码区(非翻译区);4)调控区,包括启动子和增强子等。基因编码区的两侧也称为侧翼顺序。外显子组(Exome):全部外显子称为“外显子组”(Exome)。外显子(Exon)作为真核生物基因的一部分,包含着合成蛋白质(生命活动的承担者)所需要的核心信息。外显子组约占全基因组序列的1%,大多数与疾病相关的变异位于外显子区。与全基因组测序相比,外显子组测序不仅费用较低,数据分析也更为简单,广泛应用于孟德尔遗传病、罕见综合征及复杂疾病的研究中。全外显子组测序 (Whole Exome Sequencing, WES):是指利用序列捕获技术(主要是核酸探针)将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。产品主要由Agilent等几家公司把控,不同公司的靶位点略有不同。Read(读段):高通量或二代测序平台产生的序列读取(Sequence Read)。PEread(Pair-end read),双末端测序读段;SEread(Single-end read):单末端测序读段。例如:PE250,就是读长为250bp双端测序。变异(Variation):通常指在不同个体、或同一个体的不同细胞之间,基因组或外显子组上的碱基序列的不同。研究变异的意义。变异位点作为分子遗传标记,在人类复杂疾病、动植物经济性状和育种研究及物种起源、驯化、群体历史动态等方面具有重大的指导意义。所谓指导意义,通常是“一项研究或机制研究的起点”。研究“变异”的一个哲学观或方法论,请查看聊生信之前的一篇评述类文章(点我)。单核苷酸多态性(Single Nucleotide Polymorphism, SNP):主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。多态性是群体基因组或比较基因组研究中的一个概念,有一定的发生和分布规律。因此SNP本身是针对“群体”而言的(within a population),应该在群体中占一定的比例(e.g. > 1%),即一般用来描述一个群体内不那么罕见的碱基突变。“二态性”(biallelic)。即C>T,C>G,C>A等两种“状态”之间的变异。偶尔也存在三态或四态之间的变异(需要排除测序带来的假阳性)。SNV(Single Nucleotide Variant):即单核苷酸变异,但频率没有任何限制,可用于描述任意一个可以被测序检测到的碱基突变。除了整个个体或生殖细胞,SNV也可能出现在体细胞中。体细胞的单核苷酸变异(例如肿瘤组织)也可以称为“Single Nucleotide Alteration”。对于少数变异位点的讨论,可直接使用“点突变”(Point Mutation)。SNP与SNV。二者概念的界限并不是非常明晰,日常交流时甚至一些高水平期刊上也会将这二者混用。SNP更偏向于群体研究,频率可能也较高(但又不绝对,一些SNP的频率也可能极低)。通常SNP数据库的位点总数远低于SNV。在存储SNV的数据时,应尽可能地保留所有样本的全部变异信息(如gVCF文件),这对数据的存储带来了极大的挑战,但对于寻找有意义的罕见位点的变异非常重要。利用转录组数据分析变异。事实上除了外显子组或基因组,转录组也可以鉴定SNP或SNV等变异。这就要求在设计转录组的实验方案时,小心地将“性状”、“个体”、“器官、组织或细胞”等因素也纳入,最终不仅可以获得基因表达数据,也能获得遗传变异相关的分析结果,并可继续做一些关联分析(如eQTL)。最终测序数据能反映更多的组学信息,提高研究质量。胚系突变(Germline Variant):又叫生殖细胞突变,是来源于精子或卵子的突变(父母的其它细胞也含有这样的突变),因此通常个体的所有细胞都带有突变。胚系突变可以遗传,一般用于分析遗传病。新发突变(De novo Mutations):这种突变是指父母本身没有的突变,大多是父母配子(精子、卵子)生成时产生并携带的变异,或受精卵发育过程中的自发突变。每个人身上都会有这样的变异,一般不会带来功能性的问题,但有一些先天的小儿疾病,部分新发突变刚好落在了一个重要的基因上。体细胞突变(Somatic mutation):又叫获得性突变,是体细胞(如肺、皮肤,肝脏,骨髓,眼睛等)在生长发育过程中或者环境因素影响下后天获得的突变,通常身上只有部分细胞带有突变。体细胞突变通常不会遗传给后代,通常涉及在肿瘤研究中。单有肿瘤样本时无法(直接)区分胚系突变和体细胞突变,只有加入健康样本(健康组织、血液)才能过滤掉胚系突变。克隆性造血(clonal hematopoiesis):是近几年提出的概念,指造血干细胞亚克隆所携带的突变,可能会对血液样本的WGS或WES变异检测带来一定的影响。克隆性造血的负荷与年龄的增长呈正相关,但突变丰度较低(90%的克隆性造血丰度<1%),跟来源于生殖细胞的胚系突变具有显著差异(胚系突变突变丰度一般在50%或者100%)。但对于肿瘤等体细胞突变研究影响较大,且具有个体特异性,因此必须通过同深度配对的白细胞进行过滤以及优化的生信算法等将其过滤,消除干扰。克隆性造血带有的基因变异一般是非恶性的,且对靶向治疗基因的影响很小,主要发生在DNMT3A、TET2等与靶向治疗无关的基因。插入缺失(Insertion/Deletion, InDel):是指基因组中小片段(核酸序列)的插入或缺失。结构变异(Structure Variation, SV):通常指基因组上大长度的序列变化和位置关系变化。基因组结构性变异类型很多,包括长度在50bp以上的长片段序列插入或删除、串联重复、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及复杂的嵌合性变异等。SV也可以发生在两条染色体之间,可使用Circos等软件展示。拷贝数变异(Copy Number Variation, CNV):是由基因组发生重排而导致的一种染色体结构变异,一般指长度为1 kb以上的基因组大片段的拷贝数增加或者减少(Can be as large as megabases or smaller than 1,000 base pairs), 主要表现为一个群体中的不同个体或同一个体的不同细胞之间亚显微水平的缺失和重复,即数量上与参考基因组或对照组的拷贝数不同。CNV的概念提出只是高通量短序列测序(二代测序)数据分析时,发现有些区间的覆盖度显著高于其它区间(或者是显著低于期望),但通常并不清楚他们被拷贝到了哪些地方,以及是作为整体还是被分段拷贝。CNV的分类与分布:二倍体生物的CNV可分为:正常(2拷贝)、缺失(1或0拷贝)和重复(>2拷贝)。CNV在染色体上的存在形式主要有:2条同源染色体拷贝数同时出现缺失(或同时出现重复);1条同源染色体发生缺失,另1条正常(或重复);1条同源染色体出现拷贝数重复,另1条正常。CNV的致病性。大约三分之二的人类基因组可能是由重复序列组成的,4.8-9.5%的人类基因组可以归类为拷贝数变异。CNV在人类基因组中分布广泛,是人类疾病的重要致病因素之一,可引起智力障碍、生长发育迟缓、自闭症、多种出生缺陷、白血病、肿瘤、遗传性疾病和心血管疾病等。作为疾病的一项生物标志,染色体水平的缺失、扩增等变化已成为许多疾病研究的热点。传统的方法(如FISH等)操作繁琐,分辨率和通量低,且难以提供变异区段的具体信息。CNV的来源。第一种,母源性CNV(胎儿50%可能存在相同的CNV);第二种,父源性CNV;第三种,胎儿(新发)CNV。CNV临床咨询与遗传咨询。对于母源或父源CNV,如果父母本身没有任何表型,胎儿本身也不存在超声结构异常,我们大多认为偏良性。但医生会告知由于遗传的异质性,即使胎儿的CNV来自表型正常的父母,也不能代表胎儿一定没有表型,因为有很多具有相同CNV的家系成员表型可能从无表型到严重表型。可以通过介入性产前诊断,胎儿样本做染色体核型分析和染色体微整列分析(或CNV-seq),来明确诊断CNV的大小和胎儿的具体情况(注:这里讨论的CNV大多是指临床意义不明的,对于明确致病的CNV,相应的临床建议不同)。胎儿(新发)CNV。CNV按照现有的标准分为致病性、可能致病性、临床意义不明、可能良性、良性。最难的咨询的也是临床最常见的是临床意义不明的CNV。对于新发的临床意义不明确的胎儿CNV,实际上很难咨询的,只能通过现有的数据库如decipher,OMIM,DGV等以及胎儿的超声影像学综合评估,查询CNV包含的基因情况,是否存在单倍剂量不足或三倍剂量敏感,还要结合已有的病例报道。最终的决定权还在夫妻双方,临床医生只能通过现有的资料和数据告知胎儿可能的预后情况。胎儿期的临床表型有限,只能通过超声影像评估胎儿结构方面的问题,对于出生的生长发育,智力情况无法预测。胎儿CNV的检测风险。不要轻易的决定胎儿的去与留,一定要进行介入性产前诊断明确CNV的来源、大小和致病性,并通过密切结合胎儿的超声影像学等资料进行综合评估。染色体CNV普遍存在于我们每个正常人,只是现有的知识和数据库资料有限,因此有很多临床不明的CNV难以解释。CNV相关的诊断还有很多的路要走,很多时候即使是经验丰富的临床医生也无法给出明确的临床建议。产前诊断工作如履薄冰,如临深渊,临床医生应尽最大可能给出相对准确的临床建议。InDel vs CNV。目前主流的相关分析工具(BWA,bowtie2等)和算法(Smith-Waterman的local-alignment等)能够直接鉴定出来的插入和删除(InDel),检测的范围一般是从1bp到50bp。至于更大尺度的丢失和获得,主要是通过分析序列的覆盖度鉴定为CNV。拷贝数多态性(Copy Number Polymorphism, CNP):一个CNV在群体中的频率超过1%时通常成为CNP。类似的比较可参考上文的SNP vs SNV。SD区域(Segment Duplication Region)或串联重复区域(Tandem Repeat Region):串联重复是由序列相近的一些DNA片段串联组成。例如在人类染色体22号和Y染色体上的大量SD序列。细胞周期(Cell cycle):含间期(Interphase)与分裂期(即M期:Mitosis有丝分裂;Meiosis减数分裂)两个阶段。间期分为:DNA合成前期(G1,Gap Phase 1)、DNA合成期(S,Synthesis Phase)与DNA合成后期(G2,Gap Phase 2)。分裂期分为:前期(Prophase)、中期(Metaphase)、后期(Anaphase)和末期(Telophase)。体细胞突变、癌变或生殖细胞新发突变,多发生在细胞分裂间期的S期或减数分裂的间期。G0期:在具有分裂能力的组织中,部分细胞会暂时脱离细胞周期,停止细胞分裂,即进入G0期。G0期内的细胞虽不分裂,但仍然活跃地进行代谢活动,执行特定的生物学功能。一旦得到信号指使,G0期细胞会快速返回细胞周期,分裂增殖。如结缔组织中的成纤维细胞,平时并不分裂;但一旦所在的组织部位受到伤害,成纤维细胞会马上返回细胞周期,分裂产生大量的成纤维细胞分布于伤口部位,促使伤口愈合。对G0期细胞的产生和其重返细胞周期机理的研究,已越来越受到人们的重视。不仅涉及对细胞分化和细胞增殖调控过程的探讨,而且对生物医学,如肿瘤发生和治疗、药物设计和药物筛选等,都具有重要的指导意义。全外显子测序与基因组测序的应用: 胚系(或生殖细胞)突变检测,如人类遗传病、遗传性肿瘤、植物优良表型等;体细胞突变检测,如癌症研究、寻找药物靶点、肿瘤负荷监测等。群体(Polulation):是指生活在一定空间范围内,能够相互交配并生育具有正常生殖能力后代的同种个体群。家系(Family/Pedigree):指记录某一家族各世代成员数目、亲属关系,以及有关遗传性状或遗传病在该家系中分布情况的图示。先证者(Probands):指在对某个遗传性状进行家系调查时,其家系中第一个被确诊的人。在谱系图上通常用箭头或手指图形来表示先证者。在遗传病的家系调查中最初在医院受到检查的患者就是先证者,通常每一家系中有一个人是先证者,但在检查地区内的全体人员的时候,则所有患者都是先证者。等位基因(Allele):基因的一种特殊形式。一个来自父亲,另一个相同的部分来自母亲,也有可能是新发(de novo)突变形成的新等位基因。基因型(Genotype):从个体父母那里获得的(或新发突变形成的)两个等位基因的组合。例如:GA(或G/A)。等位基因频率(Alleles Frequency, AF):在一个群体中,某类等位基因占该基因位点上全部等位基因数的比率。基因型频率(Genotype Frequence, GF):群体中某一基因型个体的数目占群体总个数的比例。遗传平衡定律或哈迪.温伯格定律(Hardy-Weinburg):在一个群体无限大,且又具备以下条件:随机交配、没有突变没有选择、没有遗传漂变的情况下,群体内一个位点上的基因型频率和基因频率将代代保持不变,处于“遗传平衡状态” ,这一平衡状态就称之为Hardy-Weinberg Equilibrium(哈迪-温伯格平衡)。连锁不平衡(Linkage Disequilibrium):相邻位点之间的非随机关联,当一个位点上的某一等位基因与另一位点上的等位基因共同出现的概率大于随机组合的假设,则这两个位点之间存在连锁不平衡。连锁平衡(Linkage equilibrium):两个基因座的等位基因组合的频率等于组成组合的等位基因各自频率的乘积,不存在优势组合,称为连锁平衡。杂合子(Heterozygous,美/hetərəˈzaɪɡəs/):具有两个不同等位基因的基因型。纯合子(Homozygous,美/hoʊməˈzaɪɡəs/ ):具有两个相同等位基因的基因型。复合杂合子(Compound heterozygous):即:(同一个基因的)双等位突变。同一个基因在每条染色体均存在变异,但发生的位置不同。复合杂合子重要意义在于:两对及以上的等位基因可以在各自杂合状态下(同时也是隐性遗传),联合起来导致疾病(无需纯合突变致,但造成与纯合突变相似的破坏性)。复合杂合度反映了许多常染色体隐性遗传疾病突变基础的多样性(diversity of the mutation base);大多数致病基因发生过多次突变。这意味着许多疾病发生在拥有两个不相关等位基因的个体身上,这些等位基因从技术上讲是杂合子(也是隐性遗传),但两个等位基因都有缺陷。表型(Phenotype):基因型和环境共同决定的基因的物理表达,如:眼睛的颜色、身高、智力、是否患病。次要等位基因(Minor allele ):其概念主要用于群体的(全基因组)关联分析。一般用A1代表minor allele,A2代表major allele,均是由实际测序样本中的分布决定。据此,可以统一基因型(Genotype)的写法,例如:A1A1,A1A2,A2A2。在群体的(全基因组)关联分析中,F_A代表minor allel在case中的频率,F_U表示minor allel在control中的频率(均是针对minor allel的定义)。例如:假设Case有基因型如下的个体:AA, AA, AT;Control有基因型如下的个体:TT, AA, TA, AA, AT, AT。故等位基因A的个数是12,T的个数为6,则A1(minor allel)为T,总占比少:6/(12+6);A2为A(major allel)。F_A=1/6≈0.1666667;F_U=5/12≈0.4166667,可见0.4166667>0.1666667,是否具有显著性则需要计算P-value,样本量少时一般选择费舍尔精确检验。先证者模式:是只对家系中第一个发现该病的患者(先证者)一个人进行检测,数据分析后有疑似致病性位点,再进行父母或其他患病成员的家系Sanger测序验证。家系trio模式:对先证者及其父母三个人(family trio)全部进行高通量测序。trio模式会提高阳性诊断率,如严重发育障碍患者,先证者模式WES阳性诊断率是28%,Trio-WES的阳性诊断率可以提高至40%。家系模式的优势之一是可进行家系表型-基因型的共分离分析,可以从遗传学上考虑到更有意义的变异位点,如:新发(De novo)突变、纯合突变、复合杂合突变、X连锁疾病半合子突变、单亲二倍体等。通过家系筛查可以判断变异致病性,有更强的遗传学证据,同时避免先证者模式可能造成的假阳性结果。 -
性别是怎么发育形成的?
胎儿性别由X、Y性染色体决定真正的男女之别,在卵子和精子结合的一刹那就注定了。卵子和精子仅拥有一半的染色体。受精后会重新组合成一个拥有23对染色体的新个体,其中,X、Y性染色体决定了宝宝性别的发育。生物课上有学过,女性只有XX染色体,男性有XY染色体,所以携带Y染色体的男性,才是决定宝宝性别的关键。古代生不出儿子不停纳妾的男性,只能怪自己不争气了。所以,宝宝的性别是由那个钻进卵子的精子所携带的性染色体决定的。胎儿是如何发育出性别的呢?这就要看性腺(卵巢或睾丸),生殖管道(输卵管、子宫或者输精管等等)的发育过程了。在宝宝发育到5~6周的时候,首先形成原始生殖腺。这时候的原始生殖腺既可以向卵巢方向,又可以向睾丸方向发育。在宝宝长到第8周时,如果携带有Y染色体,在Y染色体上的睾丸决定因子作用下,性腺就向睾丸即男性的方向发育了;若没有Y染色体,就向卵巢的方向发育了,成为女宝宝。啥时候才能知道宝宝的性别?我们肉眼能识别的性别差异是外生殖器。男宝宝和女宝宝初显差别最早在12周;到16周时,才能通过外生殖器确定胎儿的性别。然而,如若是想通过无创方法了解宝宝的性别,可能还要略晚一些。约20周以后,才能通过超声等无创性措施才可初步判断宝宝的性别。在某些特殊情况下,伴随性染色体遗传的疾病如色盲等等,可能需要通过性别鉴定来减少疾病的发生,未必需要等到20周以后,可以早期通过科学技术来实现。常用的如胚胎种植前遗传学诊断、绒毛组织、羊水的检测等等,但是多数技术是有创伤的,可能影响准妈妈和宝宝的健康,一般不推荐使用。生男生女可以人为决定吗?关于影响生男生女的因素众说纷纭,最普遍的说法是酸碱体质影响孩子的性别。在碱性环境中,含“Y”染色体的精子比较活跃,容易优先受精,形成男宝宝,因此有人认为多吃碱性食物可增加生男宝宝的概率。同样道理,有人认为用碱性溶液如苏打水冲洗阴道,通过改变阴道酸碱度也有利于生男宝宝。当然,这也已经被辟谣了。不冲洗出阴道炎已经万幸了。除此之外还有其他说法,如从事消耗能量较高的工作或运动的男性,肌肉及体液中积累的酸性代谢物质过多,可影响含“Y”染色体精子的活力,从而降低生男宝宝的概率,等等。关于生男生女的说法很多,基本都围绕着酸性和碱性环境的变化。虽说确有科学证据表明,“Y”染色体在碱性环境下更容易存活,但其差别非常微弱,想通过这点操控孩子性别是非常困难的。更何况人体为了维持正常生理活动,有自动调节体内酸碱平衡的能力。从女性健康角度来说,正常阴道菌群会维持酸性环境,以此来抑制病原体生长,称为阴道自净作用。随意改变阴道的酸碱性,会导致女性患上阴道炎等疾病,甚至影响受孕。随着医学研究的发展,目前人们在孕前、孕早期和孕中期通过活检胚胎上的部分细胞、取绒毛细胞或从羊膜腔穿刺抽取羊水等,经特殊技术处理后,可以识别染色体类型,以此来鉴定胎儿性别。但是这些技术仅仅适用于携带遗传疾病的人,绝大多数国家,包括中国,都严令禁止非治疗目的的性别选择。研发这些技术的目的不是为了迎合人们生男生女的意愿,而是为优生考虑。由于某些疾病, 如血友病及假肥大型进行性肌营养不良等致病基因在“X”染色体上,而男性只有一条“X”染色体,因此男孩发病率高。鉴于这些疾病严重危害人们的健康,可以通过种植前遗传学诊断技术,在妊娠前选择女性胚胎移植到子宫内,以此期望获得女性婴儿而减少这些遗传性疾病的发生。也就是第三代试管婴儿技术。这种技术可以对胚胎性别进行准确判断。三代试管谁都可以做吗?在国内,如果是非医学需要,却通过第三代试管婴儿来生男或生女是被明令禁止的!第三代试管婴儿是指胚胎植入前染色体筛查或基因诊断,它可以筛查出有遗传疾病的胚胎,从而在较大程度上保障所植入胚胎的健康。第三代试管的适应症包含:染色体数目或结构异常的患者;夫妻一方为性连锁遗传病的携带者(例如血友病、假肥大性肌营养不良);可进行基因诊断的单基因病患者或者携带者等。由此可见,优生优育才是第三代试管婴儿的目的。而确定胚胎性别,只是其附带的“福利”。虽然通过第三代试管婴儿生男孩或女孩不可取,但对大龄夫妻、性连锁遗传病患者或相关基因的携带者及部分单基因遗传病患者等不孕不育人群而言,第三代试管婴儿确实是实现优生优育的重要手段。现实生活中,生男生女的概率虽不是绝对的一比一,但大体均等,这是自然的规律。无论男孩、女孩,都是大自然赐予人类的最好礼物,而人为干预婴儿性别,既违反了法律,也会破坏自然的平衡。我们应当谨记,对宝宝的性别我们无法选择,也不必选择,自然的就是最好的。 -

什么是出生缺陷?
出生缺陷是婴儿出生前发生的身体结构、功能或代谢方面的异常,是导致早期流产、死胎、婴幼儿死亡和先天残疾的主要原因。比如先天结构畸形、染色体异常、遗传代谢性疾病、盲、聋、智力障碍等等功能异常都属于出生缺陷。
我国出生缺陷的发生率高吗?
据估算,我国出生缺陷的发生率是5.6%,以全国年出生数1600万计算,每年新增出生缺陷约90万例,其中出生时有临床明显可见的出生缺陷约25万例。出生缺陷有哪些危害?首先出生缺陷导致健康危害,它是导致婴儿死亡的首要原因,出生缺陷存活儿中,约5%在1岁前死亡;出生缺陷也导致儿童各类疾病发生率和残疾率增加。其次出生缺陷对家庭来说,会严重扰乱家庭成员的正常生活和工作,带来沉重的经济压力以及巨大的精神压力和精神负担。第三对社会来说,需要提供更多相应的社会保障和支持、康复教育服务等,可以使劳动生产力下降,降低了人群健康水平和人口素质。
引起出生缺陷的原因是什么呢?
遗传性因素和环境因素均可导致出生缺陷发生。其中遗传因素占了40%,包括有单基因遗传、多基因遗传、染色体病等,环境因素占5-10%,包括化学、物理、生物及营养因素等等。还有50%是遗传和环境两种因素共同作用或其他不明原因导致。
怎样预防出生缺陷发生呢?
由于出生缺陷危害巨大,政府高度重视出生缺陷防治工作,大力推广三级预防措施。一级预防是预防缺陷的发生,包括健康教育、婚前医学检查、孕前保健、遗传咨询、最佳生育年龄选择、增补叶酸等措施;二级预防是对已经发生的缺陷在孕期早发现、早诊断、早采取措施,以减少严重缺陷儿出生,产前筛查产前诊断就是二级预防的重要措施;三级预防是对已经出生的缺陷患儿采取及时有效的诊断、治疗和康复,防止病残,提高患儿生活质量,比如现在广泛开展的新生儿足跟血进行遗传代谢病的筛查还有听力筛查、先心病筛查等。
什么是产前筛查、产前诊断?
绝大多数出生缺陷在出生前没有明显的表现,常规的产检无法发现,只能通过专门的检查发现。产前筛查是指通过简单、经济、无创的方法在普通的孕妇群体中发现可能怀有异常胎儿的高风险孕妇,对筛查出的高风险孕妇再实施诊断性检查,也就是产前诊断。产前诊断是指对胎儿进行先天性缺陷和遗传性疾病的诊断。
目前产前筛查主要针对的疾病是哪些?
目前产前筛查主要针对的疾病有染色体病、严重结构畸形、神经管缺陷。筛查方法都是无创伤性的,有抽血进行唐氏筛查或者无创DNA产前筛查,有超声筛查;产前诊断则包括有创性产前诊断和无创性产前诊断,有创的产前诊断即绒毛活检、羊水穿刺、脐带血穿刺,通过抽取样本进行细胞遗传、分子遗传以及生化检测等,主要进行胎儿染色体病的检查,无创产前诊断则是指影像学检查,最常用的就是超声,此外还有核磁共振,主要是进行结构畸形的诊断。检查时间一般唐氏筛查是孕15-20+6周,无创DNA是孕12-22+6周,超声有孕11-13+6周的NT检查,孕20-24周的大排畸。
父母都健康,为什么还要进行筛查?
所有的孕妇都有可能生育出生缺陷儿。以唐氏综合征为例,随着孕妇年龄增加疾病发生率逐渐增加,患儿表现主要是智力障碍,生活不能自理,一辈子要别人照顾,一部分唐氏儿还可能合并有先天畸形,如先天性心脏病、听力及视力障碍等,到目前为止,还没有有效的治疗措施,产前筛查产前诊断是目前最有效的预防手段,理论上讲,不管是否有家族史,正常人也有可能生育唐氏综合征胎儿。所以建议所有的孕妇都有必要进行产前筛查,必要时实施产前诊断。
产前筛查异常,是不是胎儿一定有问题?
要注意筛查不是诊断,筛查结果异常只是提示有一定的患病风险,并不表示胎儿一定异常,需要实施产前诊断来进一步确诊,临床上也不会因为筛查高风险而终止妊娠。
产前诊断适用哪些孕妇呢?
所有的孕妇都需筛查,只有存在某些高危因素的孕妇才建议产前诊断,高危因素有:1.羊水过多或者过少;2.胎儿发育异常或者胎儿有可疑畸形;3.孕早期接触过可能导致胎儿先天缺陷物质;4.夫妇一方患有先天性疾病或遗传性疾病,或有遗传病家族史;5.曾经分娩过先天性严重缺陷婴儿;6.孕妇预产期年龄达到或超过35周岁;7.有两次以上不明原因的流产、死胎或新生儿死亡者;8.产前筛查结果异常者;9.医师认为有必要进行产前诊断的其他情形。存在以上情况的,建议产前诊断。
-

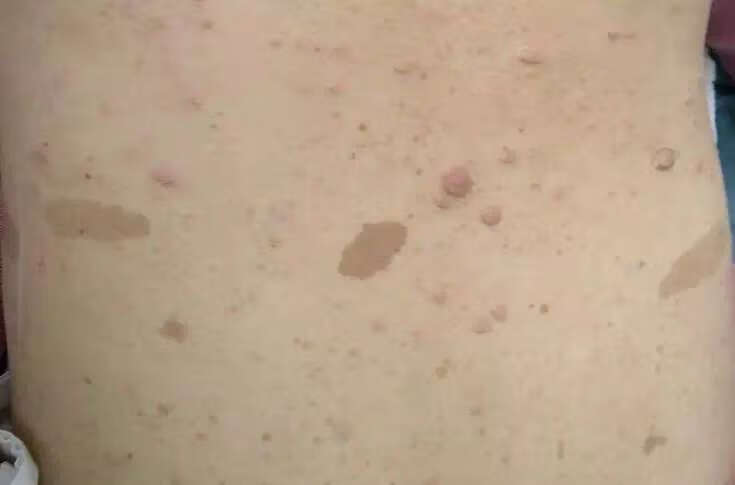
最近接诊了不少NF1基因突变的病人,有的表现为巨大的头面部神经纤维瘤,有的仅仅表现为牛奶咖啡癍。随着基因检测技术的发展,越来越多有“胎记”的小朋友捡出了NF1基因突变,那么小孩罹患神经纤维瘤吗?孩子还这么小,今后该怎么办呢?
为了给家长给出明确的咨询意见,让“临床意义不明”明确起来,让家长不再抓盲,特意查阅相关文献资料,做出以下科普文章,仅供大家参考讨论。牛奶咖啡斑为遗传性皮肤病,本病色素斑处的黑素细胞和角质形成细胞内黑素增多,黑素细胞活性亢进,产生大量黑素,形成色素沉着斑。它产生的原因有很多,因人而异。牛奶咖啡斑为淡褐色斑,棕褐色至暗褐色,大小不一,圆形、卵圆形或形状不规则,边界清楚,表面光滑。牛奶咖啡斑的病理改变有表皮黑素增加,特别见于基底层中,多巴染色黑素细胞及基底层的角质形成细胞中有巨大黑素体,基底层黑素细胞正常或略有增加。

神经纤维瘤病是一种由于基因突变导致的以皮肤牛奶咖啡斑、多发性神经纤维瘤或听神经瘤等多系统损害为特征的常染色体显性遗传病。由于基因缺陷使得神经嵴细胞发育异常导致的一类多系统损害疾病,以生长在神经系统的良性肿瘤为主要表现。其发病率约1/3000,每3000个出生婴儿中约有1个患儿,无明显男女差异,有家族聚集和遗传倾向。保守估计目前我国神经纤维瘤病患者80-100万,每年新增4000-5000例。
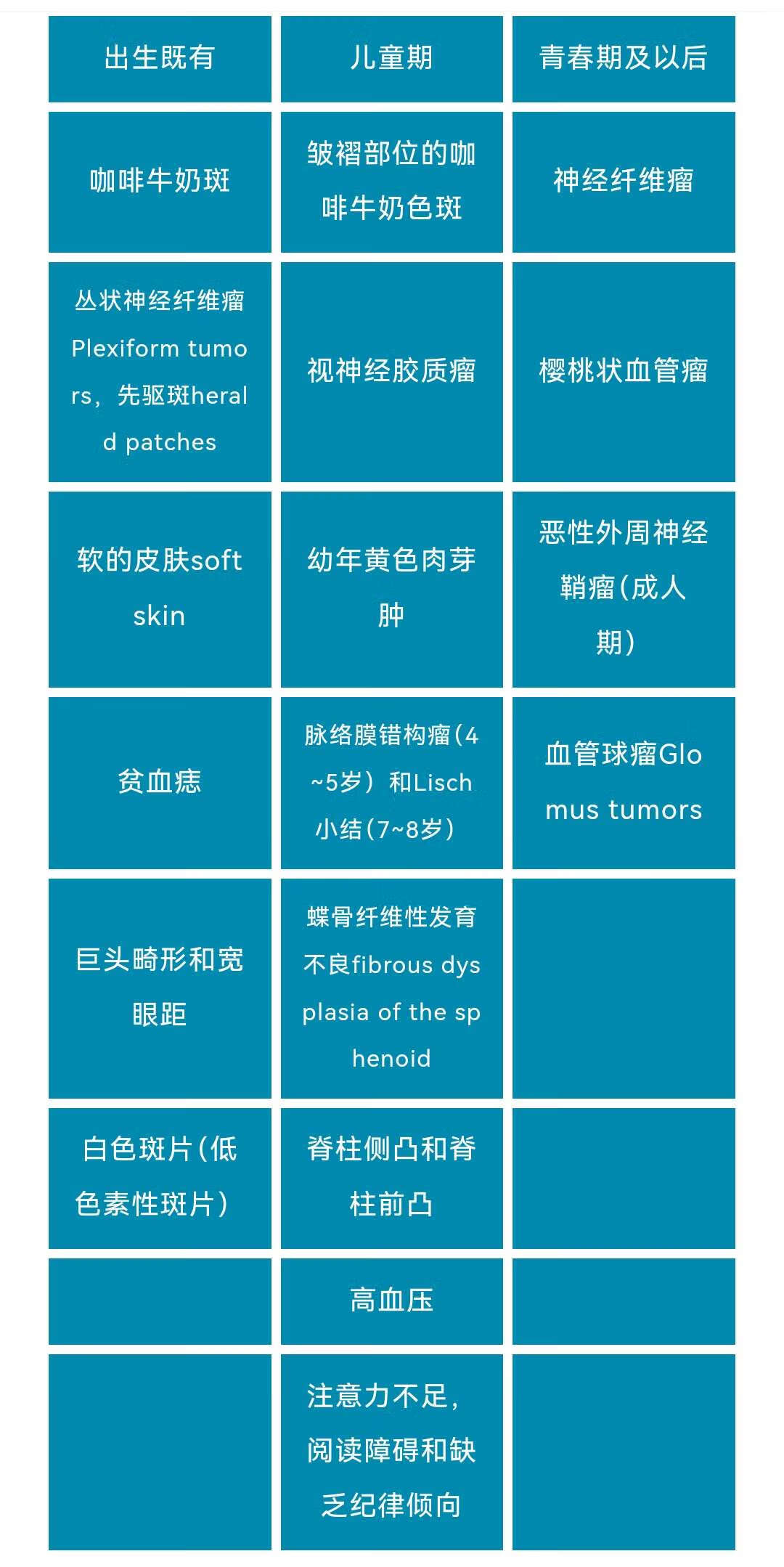
因,定位于17号染色体。但NF1突变的临床异质性很大,即在病人之间表现差异会很大,许多患者仅仅表现非常轻微的可能仅有皮肤牛奶咖啡斑的问题;约1/3的病人一生中可能会出现一种或多种并发症,这些并发症中一部分是比较轻的易于治疗的问题,一部分可能会是很严重的问题。各常见体征的起病年龄如下表:
具体可能出现的各项体征下面逐一介绍,首先可大致分为两大块,一块为皮肤体征,一块为皮肤外体征,皮肤外体征又可具体往下细分为眼部症状、骨科症状、肿瘤、血管异常、神经系统体征。下面的图片均引用自Gianluca Tadini主编的Atlas of Genodermatoses第二版一书。在本篇科普的最后一部分将介绍后期如果随访。



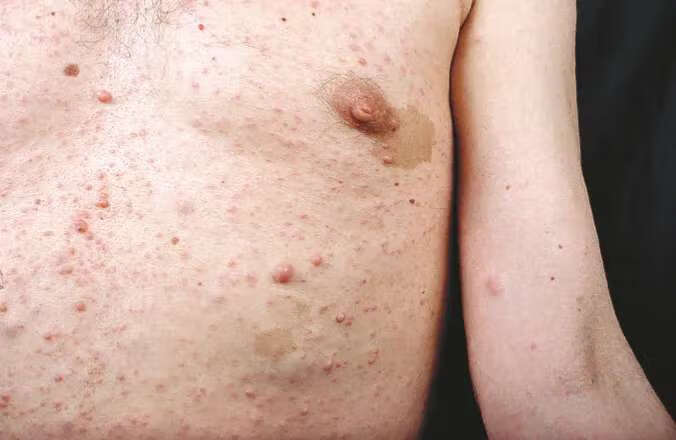
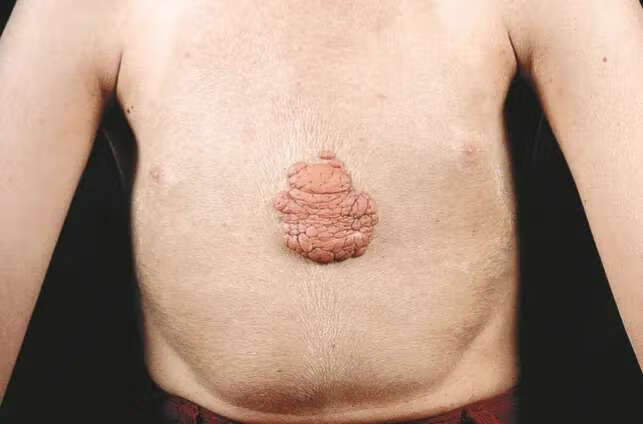

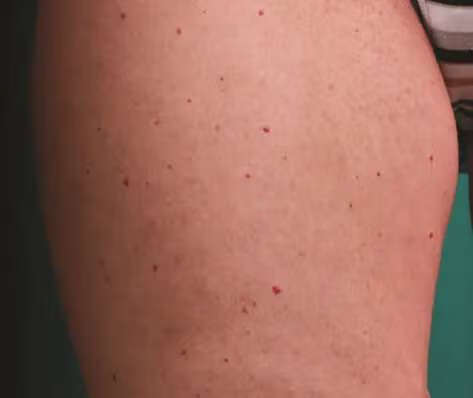


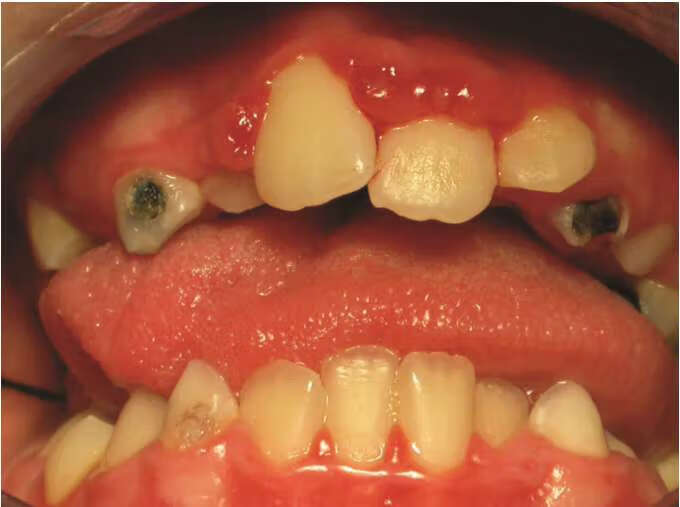


非皮肤症状
一、眼科体征(注:每年一次视力检查直至7~8岁,可能需要用到一些特殊检查办法)


二、骨骼:
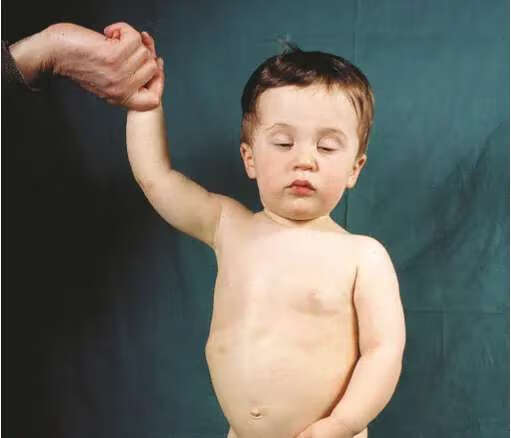

三、肿瘤形成:

四、血管异常:
早发性的高血压
肺动脉狭窄(这种关联称为Watson综合征)
系统性血管异常,尤其是肾动脉和脑动脉烟雾病样型脑动脉(罕见)
五、神经系统体征:丛状神经纤维瘤通常条索状分布,可引起压迫症状 约10%的患者出现癫痫(由于脑补错构瘤)
多发性硬化症智力低下可以出现,但其实很少。通常都是由于这些患者因在学校里表现不好,被误认为是智力低下
入学的前几年,注意力不集中、诵读困难、缺乏纪律性的情况可能很严重
-

随着基因测序技术和生物信息学的快速发展,个体化精准医疗的发展成为热点,也将为大众带来福利。然而,遗传性疾病以及基因检测技术专业知识和民众之间存在巨大的信息鸿沟,遗传咨询师将发挥重要的桥梁作用。
何为遗传咨询
遗传咨询(genetic counseling)是帮助人们理解和适应遗传因素对疾病的作用以 及其对医学、心理和家庭的影响的程序。(美国国家遗传咨询协会,NSGC,2006.5)
风险评估/专业支持/技术支持/沟通技巧:① 通过对家庭史的解释来评估疾病的发生和再发风险率;
② 进行有关疾病的遗传、实验室检测、治疗处理及预防的教育,并提供与疾病有关的各种可以求助的渠道及研究方向;
③ 辅导促进知情选择和对所患疾病及其再发风险的逐步认知和接受。
遗传咨询内容
遗传病知识介绍
1)遗传病基本情况:包括病因、流行病学、遗传方式、症状和体征
2)遗传病对策:a.诊断 b.治疗 c.预防
风险评估
1)单基因病再发风险率:父母基因型已推定/未推定时再发风险率的估算
2)染色体病和多基因病以其群众发病率为经验危险率
健康指导
综合分析病情、家族史、临床辅助资料、基因检测结果,给予饮食、运动等生活习惯的建议;推荐相关专科就诊;长期随访
遗传咨询发展历程


遗传咨询医师岗位职责
遗传咨询医师是遗传健康保健队伍中的一员,起到沟通病人 与其他各类不同的医学遗传专业人员(医生)之间桥梁作用。

遗传咨询医师的职业技能
1 获得并解释个人和家庭的病史、发育史与生育史
2 分析出遗传方式以及遗传疾病和先天缺陷的发生风险与再发生风险
3 解释遗传疾病的病因、病史、诊断与应对措施
4 说明并解释基因检测结果与其它诊断依据;
5 使用心理评估识别情感、社会、教育以及文化问题
6 评测出客户和/或家庭对出现疾病或存在疾病发生风险的反应程度
7 以病人为中心进行服务并进行先期引导 促进客户在充分了解情况的基础上做出有关检测、临床干预、生育以及与家庭成员进行沟通的决策
8 发现并使用能提供医学、教育、经济以及心理方面支持的社区资源 为家庭以及其它健康医务专业人员提供有关医药、遗传与咨询方面信息的书面文件
遗传咨询医师必备的临床技能
1、正确解读临床基因检测结果
2、熟悉表型与基因之间的复杂关系
3、熟悉表型特异性&诊断潜力
4、熟练查询人群数据库 (表型正常人及疾病)
5、熟练使用常用染色体CNV数据库、频率数据库,其他常用资源
6、熟悉常用的位点预测软件、解读规则 、报告规范标准
-
 后背疼痛难忍是什么情况
后背疼痛难忍是什么情况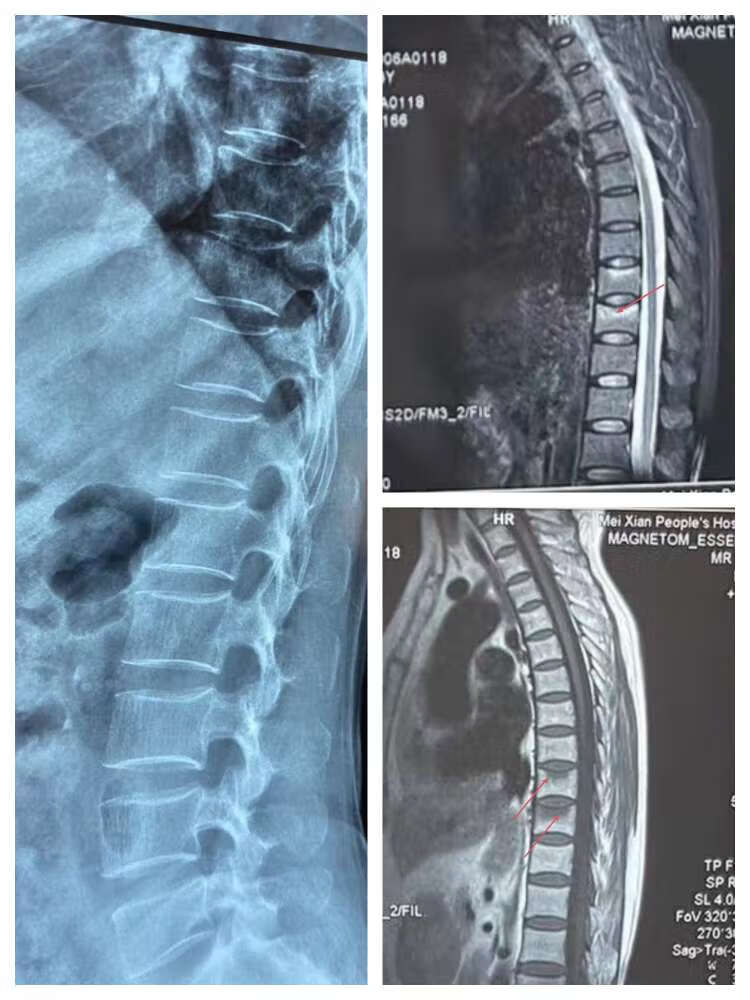 图示为疼痛科今天就诊的一位63岁女性患者,近半年一直照顾高龄老母亲,近期老母亲摔倒,抱扶老母亲后出现背痛难忍,在外院拍片正常,口服药物效果不佳,连弯腰翻身都困难,这是个什么情况?问诊查体,考胸椎骨折,行胸椎核磁共振检查,“凶手”浮现,如图所示可见胸9和胸10两个椎体骨折。为什么会骨折?考虑是骨质疏松征所致!有效的抗骨质疏松治疗+晒太阳+运动+营养可以很快改善症状、防治骨折和改善生活质量。
图示为疼痛科今天就诊的一位63岁女性患者,近半年一直照顾高龄老母亲,近期老母亲摔倒,抱扶老母亲后出现背痛难忍,在外院拍片正常,口服药物效果不佳,连弯腰翻身都困难,这是个什么情况?问诊查体,考胸椎骨折,行胸椎核磁共振检查,“凶手”浮现,如图所示可见胸9和胸10两个椎体骨折。为什么会骨折?考虑是骨质疏松征所致!有效的抗骨质疏松治疗+晒太阳+运动+营养可以很快改善症状、防治骨折和改善生活质量。 -

#甲状腺相关疾病不要频繁按摩挤压!#甲状腺结节 #动画演示 #医学科普 #甲状腺医生李德志 #医者仁心 @抖音小助手
-

#黄斑变性 #视物变形 #医学科普 #眼科医生聂红平
展开更多


营业执照 Investor Relations
违法和不良信息举报电话:4006561155 消费者维权热线:4006067733
医疗监督热线:950619(京东),0951-12320(宁夏),0951-12345(银川)
Copyright © 2022 jd.com 版权所有

